Досягнення в розумінні та лікуванні діабетичної хвороби нирок: фокус на механізмах тубулоінтерстиціального запалення
Дата публікації: 31.10.2023
Автори: Відкриті джерела , Редакція платформи «Аксемедін»
Ключові слова: діабетична хвороба нирок, епітелій, канальці, фільтрація, інтерлейкін
Цукровий діабет є глобальним хронічним захворюванням, захворюваність на яке постійно зростає. Останні дані Міжнародної діабетичної федерації (IDF) передбачають, що до 2045 року понад 10,5% дорослих людей у всьому світі будуть хворіти на діабет, що призведе до ураження нирок у 100-250 мільйонів людей. Раніше панувала думка, що діабет, особливо діабет 2 типу, переважно вражає клубочкові структури нирок, стан, який називають діабетичною нефропатією (ДН). Однак, оскільки наше розуміння патогенезу, клінічних проявів і патологічних моделей ураження нирок при цукровому діабеті покращилося, у рекомендаціях Національної ниркової фундації з дослідження якості результатів захворювання нирок (NKF/KDOQI) у 2007 році було введено термін «діабетична хвороба нирок» замість діабетичної нефропатії. Tervaert та його колеги повідомили у 2010 році, що деякі особи з цукровим діабетом і ураженням нирок можуть не мати клінічних ознак альбумінурії. Однак у цих пацієнтів спостерігається порушення функції нирок та/або клінічні прояви канальцевої дисфункції, такі як нирковий канальцевий ацидоз.. Результати біопсії нирок показали переважне ураження канальців, інтерстицію та/або кровоносних судин, що супроводжується відносно нехарактерними змінами клубочків, такими як потовщення базальних мембран клубочків, розширення мезангіального матриксу та гломерулосклероз. Подальші дослідження підтвердили ці патологічні висновки. Навіть у пацієнтів з DKD, які переважно проявляють ураження клубочків, біопсія нирки виявила різні ступені тубулоінтерстиціального ураження. Ці висновки спонукали Американську діабетичну асоціацію (ADA) та NKF до консенсусу змінити термінологію з «діабетичної нефропатії» на «діабетична хвороба нирок» з наміром більш точно описати спектр порушень функції нирок, спричинених діабетом. Ця зміна підкреслює розвиток розуміння та визнання складної ролі ураження нирок у контексті діабету.
Рекомендуємо долучитись до семінару «Незвична комбінація — очевидна перевага та ефективність» про найсучасніші комбінації препаратів для лікування цукрового діабету.
Раніше вважалося, що DKD головним чином включає патологічні зміни клубочків. Однак нещодавні дослідження виявили тісний зв’язок між ступенем тубулоінтерстиціального ураження та прогресуванням функції нирок, а також прогнозом. Тубулоінтерстиціальні ураження можуть виникати незалежно від ураження клубочків. Клінічні дослідження припустили, що зниження швидкості клубочкової фільтрації (ШКФ) у пацієнтів з DKD без протеїнурії в основному пояснюється тубулоінтерстиціальним пошкодженням. Активація судинного ендотеліального фактора росту (VEGF) у поєднанні зі зниженим рівнем оксиду азоту (NO) може викликати вазоконстрикцію в малих кровоносних судинах. Підвищений артеріальний тиск і субоптимальний контроль рівня глюкози в крові, що призводить до механічного стресу, сприяє зниженню перитубулярного кровотоку, посилюючи гіпоксію. Ці висновки в сукупності свідчать про те, що пошкодження клітин проксимального канальцевого епітелію (PTECs) не тільки впливає на їх функціонування, але також поширюється на більш значні пошкодження клубочків, охоплюючи пошкодження подоцитів. Отже, пошкодження канальців може виникнути до пошкодження клубочків, що вимагає подальшого дослідження.
Точні механізми, що лежать в основі тубулоінтерстиціальних уражень при DKD, до кінця не вивчені. Однак імунне запалення визнано характерною ознакою, пов’язаною з розвитком і прогресуванням ушкодження канальців при DKD. На ранніх стадіях захворювання спостерігаються гіпертрофія та збільшення кількості клітин епітелію канальців, а також потовщення канальцевої базальної мембрани. Вважається, що ці фактори мають вирішальне значення для ініціювання та сприяння процесу тубулоінтерстиціального фіброзу. Тривала присутність високих рівнів глюкози в крові разом з ішемією та гіпоксією призводить до апоптозу канальцевих клітин, атрофії канальців та дегенерації. Запальна клітинна інфільтрація, збільшення продукції факторів запалення, інтерстиціальний фіброз, артеріосклероз інтерстиціальних дрібних артерій та гіалінові зміни в артеріолах також сприяють виникненню та прогресуванню уражень.
Реєструйтесь на майбутні семінари в межах Nephro Wednesday, або перегляньте записи тих, що вже завершились.
Існуючі методи лікування, такі як контроль глікемії, блокада ренін-ангіотензин-альдостеронової системи, стабілізація гемодинаміки, а також профілактика та лікування ускладнень, не здатні повністю пригнічувати прогресування DKD, що підкреслює нагальну потребу в нових терапевтичних цілях. За останнє десятиліття, крім інгібіторів SGLT-2, антагоністи мінералокортикоїдних рецепторів (MRA) також продемонстрували багатообіцяючі результати як клас препаратів. Нове покоління нестероїдних селективних МРА, фінеренон, може запобігти різним ушкодженням, спричиненим надмірною активацією МР, запальним захворюванням і процесам фіброзу, тим самим сприяючи відновленню функції серця та нирок. З 2021 року фінренон був схвалений FDA, Європейським агентством з лікарських засобів (EMA) і Китайським національним управлінням медичної продукції (NMPA) для використання у пацієнтів із ДЗН і пацієнтів із недіабетичним захворюванням нирок (NDKD), що супроводжується хронічним запаленням та інтерстиціальним фіброзом.
Поглиблене вивчення імунозапальних механізмів, що лежать в основі тубулоінтерстиціальних уражень при ДЗЗ, і потенційних цільових методів лікування має велике клінічне значення для лікування ДЗЗ. У цьому огляді обговорюватимуться імунозапальні механізми тубулоінтерстиціальних уражень, спричинених діабетом, та їхній зв’язок із потенційною таргетною терапією. Крім того, в огляді буде надано детальний огляд патогенезу ураження канальців при DKD та використання ліків, щоб запропонувати нові ідеї для розробки стратегій лікування DKD.
Механізми ушкодження ниркових канальців при ДЗН
Ниркові канальці та інтерстицій становлять понад 90% об’єму нирки та відіграють важливу роль у таких функціях, як реабсорбція, секреція та екскреція. Серед цих функцій проксимальні епітеліальні клітини ниркових канальців відіграють вирішальну роль у реабсорбції майже всієї відфільтрованої глюкози, білків і електролітів із клубочка. Адекватне кровопостачання та доставка кисню мають важливе значення для підтримки їх належної функціональності. Порушення ниркових канальців може призвести до витоку таких речовин, як глюкоза, амінокислоти та білки, у сечу, що призводить до порушень концентрації та розведення сечі, електролітного дисбалансу та порушень кислотно-лужної рівноваги, серед інших клінічних проявів. Деякі дослідження припускають, що початкова причина мікроальбумінурії при DKD може походити від зниженої здатності до реабсорбції альбуміну епітеліальних клітин проксимальних канальців нирок, а не від пошкодження клубочків. У цьому огляді наша основна увага зосереджена на запальних механізмах, пов’язаних з ураженням ниркових канальців. Початок стійких високих рівнів глюкози (ГГ) запускає каскад подій із залученням запальних клітин і факторів, що призводить до активації поверхневих рецепторів на епітеліальних клітинах ниркових канальців і, таким чином, прискорює прогресування ДЗН.
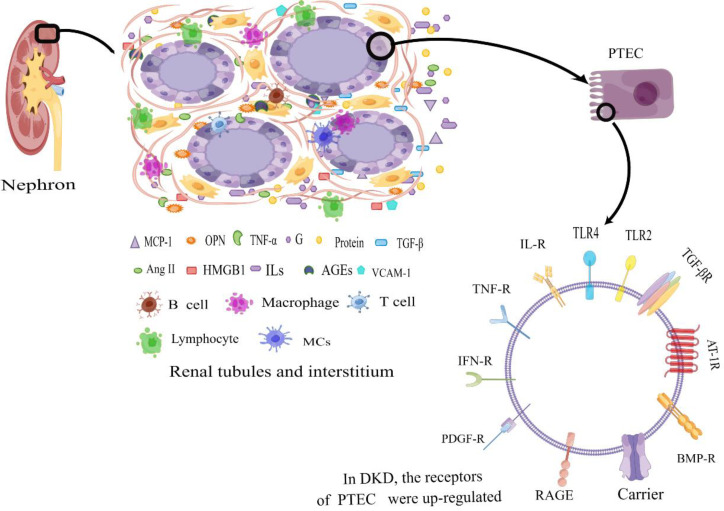
Механізми метаболічних порушень
Гіперглікемія
Гіперглікемія діє як ініціюючий фактор, що спричиняє тубулоінтерстиціальне пошкодження при DKD. На ранніх стадіях DKD у відповідь на гіперглікемічне середовище епітеліальні клітини канальців зазнають проліферації, гіпертрофії та надмірної реабсорбції глюкози та натрію. Це порушення нормальної функції призводить до дисрегуляції тубулогломерулярного механізму зворотного зв’язку, що призводить до високого внутрішньоклубочкового тиску, посилення перфузії та підвищення швидкості фільтрації. Крім того, підвищений рівень глюкози сприяє запальним реакціям. Прямий вплив високого рівня глюкози на клітини ниркових канальців сприяє виробленню прозапальних факторів, включаючи молекули внутрішньоклітинної адгезії, такі як молекула міжклітинної адгезії 1 (ICAM-1), хемокіни, такі як моноцитарний хемоаттрактантний білок-1 (MCP-1) і остеопонтин (OPN). Ці прозапальні фактори приваблюють запальні клітини, сприяючи їх інфільтрації в нирковий тубулоінтерстицій. Це, своєю чергою, викликає вивільнення низки медіаторів запалення, що призводить до пошкодження ниркової тканини та бере активну участь у прогресуванні DKD. Примітно, що серед цих факторів OPN виступає як ключовий хемокін, відповідальний за індукцію інфільтрації моноцитів/макрофагів у тубулоінтерстицій. Патогістологічне дослідження біоптатів нирок у осіб із ДЗН послідовно демонструє поширеність макрофагів серед інфільтруючих лейкоцитів у діабетичних нирках. Агрегація макрофагів усередині тубулоінтерстицію є значущим прогностичним маркером, що відображає тяжкість ниркової недостатності та ступінь інтерстиціального фіброзу. Варто зазначити, що накопичення макрофагів в інтерстиціальному просторі, а не в клубочках, тісно пов’язане з протеїнурією та прогресуючим зниженням функції нирок. Крім того, ступінь інтерстиціальної інфільтрації пропорційно корелює зі швидкістю погіршення функції нирок.
Дослідження продемонстрували посилення експресії CD40 та CD40L як у канальцевих епітеліальних клітинах, так і в макрофагах у контексті DKD. Це явище було підтверджено результатами патології ниркової біопсії та клітинними експериментами. За наявності гіпоксичних станів експресія CD40 в епітеліальних клітинах канальців запускає секрецію інтерлейкіну 6 (IL-6), тим самим полегшуючи взаємодію між запальними клітинами всередині ниркового інтерстицію та ниркових канальців. Примітно, що передача сигналу CD40 в макрофагах посилює вироблення прозапальних і профіброзних медіаторів, сприяючи пошкодженню канальців. Після стимуляції рецептори CD40 залучають різні фактори, пов’язані з рецептором фактора некрозу пухлини (TRAF), ініціюючи каскади передачі сигналів, які охоплюють фосфоінозитид-3-кіназу (PI3K), ядерний фактор каппа-B (NF-κB) і кіназу, регульовану позаклітинним сигналом p38. Сигнальні молекули, активовані CD40, мають подвійну природу, впливаючи як на прозапальну, так і на протизапальну відповіді. Заміна специфічних пептидів у CD40L викликає селективні модифікації сигнальної та ефекторної здатності CD40. Це призводить до індукції цитокінів і експресії ферментів, сприяючи запальним реакціям. Одночасно це перетворення сигналу може підвищити синтез протизапальних цитокінів. Виразні функціональні зміни, що спостерігаються в залишках макрофагів, підкреслюють їхню плейотропну роль у цьому контексті. Однак точні механізми дії CD40 в середовищі DKD, включаючи специфічні шляхи, які він залучає, залишаються не повністю вивченими і вимагають подальших досліджень. Глибше розуміння ролі CD40 у патогенезі DKD має потенціал для виявлення важливих терапевтичних цілей.
Щоб адаптуватися до середовища HG, поверхневі рецептори клітин ниркових канальців активізуються. Зокрема, у культивованих PTEC вплив рівнів HG викликає експресію toll-подібного рецептора 4 (TLR 4) і запускає вивільнення ендогенного TLR-ліганду високомобільної групи box-1 (HMGB-1) з канальцевих епітеліальних клітин і подоцитів, опосередкований активацією протеїнкінази С (PKC). Ця комбінація факторів завершується активацією NF-κB і подальшою активацією експресії IL-6 і хемокінового ліганду 2 (CCL-2)/MCP-1). Примітно, що TLR4 демонструє підвищену експресію в ниркових канальцях, демонструючи позитивну кореляцію з інтерстиціальною макрофагальною інфільтрацією та рівнями глікованого гемоглобіну (HbA1c), одночасно демонструючи негативну кореляцію з оціненою ШКФ, визначеною під час біопсії нирки. Крім того, TLR4-опосередкований шлях продемонстрував потенціал для сприяння тубулоінтерстиціального запалення при DKD. Подібним чином, TLR2 був причетний до стимуляції збільшення виробництва прозапальних цитокінів через шляхи мітоген-активованої протеїнкінази (MAPK), P38 і ERK у різних контекстах захворювань. У контексті стрептозотоцин-індукованого діабету щурів, канальцева експресія TLR2 свідчить про значне посилення регуляції в поєднанні з підвищеною нирковою експресією MCP-1 і факторів мієлоїдної диференціації, таких як первинна відповідь мієлоїдної диференціації 88 (MyD88). Це супроводжується активацією NF-κB, інфільтрацією макрофагів і наявністю ендогенних лігандів, таких як білок теплового шоку 70 (HSP70) і HMGB1, які взаємодіють з TLR. Примітно, що активація TLR2, TLR4 і специфічного рецептора кінцевого продукту розширеного глікозилювання (RAGE) служить ключовим механізмом, за допомогою якого пов’язані з пошкодженням молекулярні структури (DAMP) сприяють імуноопосередкованому пошкодженню при ДЗН.
Рекомендуємо переглянути запис семінару «Цукровий діабет, дисліпідемія і серцево-судинні ускладнення: шляхи вирішення проблеми» від провідного спікера Геннадія Кочуєва!
Традиційно визнано, що гіперглікемія індукує глікозилювання регуляторних білків комплементу, згодом порушуючи їхню регуляторну здатність. Цікаво, що блокада комплементу або активності MCP-1 показала ефективність у полегшенні тубулоінтерстиціального пошкодження. Отже, припускають, що пошкодження канальцевих епітеліальних клітин може посилюватися підвищеними концентраціями активуючих компонентів у системі комплементу та пов’язаних з ним ендогенних регуляторних білків (таких як C3, C3a, C5a, C5b–9 та Crry). Крім того, певні компоненти системи комплементу мають хемотаксичні властивості, таким чином потенційно викликаючи запальну реакцію, яка завдає пошкодження тубулоінтерстицію. Вивільнення біологічно активних речовин, у тому числі трипсину, хімотрипсину, трансформуючого фактора росту-β1 (TGF-β1), реніну та фактора некрозу пухлини-альфа (TNF-α), у тубулоінтерстицій через дегрануляцію тучних клітин ще більше підсилює запалення нирок. Крім того, шлях комплементу (C3a) і шлях TLR, пов’язані з мікробними рецепторами розпізнавання паттернів, є додатковими каналами, через які може бути спровоковано запалення нирок.
Підвищений рівень глюкози відіграє ключову роль у сприянні тубулярного та інтерстиціального фіброзу. Під впливом ГГ та інших стимулюючих факторів PTEC зазнають фенотипічної трансформації, приймаючи міофібробластоподібний фенотип у межах тубулоінтерстицію. Одночасно ці клітини виробляють профіброзні фактори, такі як TGF-β1, який сприяє синтезу позаклітинного матриксу (ECM) і посилює пошкодження нирок. Слід зазначити, що TGF-β є ключовим цитокіном у стимулюванні фенотипічної трансформації епітеліальних клітин, а інші цитокіни, такі як інтерлейкін-1 (IL-1), регулюють продукцію TGF-β (59). TGF-β1 стимулює збільшення синтезу різних компонентів ECM, включаючи ламін, фібронектин (FN) і колаген типу IV, що призводить до надмірного виробництва ECM. Крім того, TGF-β1 перешкоджає експресії та активності матриксних металопротеїназ (MMP), які є ключовими для деградації ECM, одночасно збільшуючи експресію та активність інгібіторів MMP. Отже, ці дії разом сприяють розвитку тубулоінтерстиціального фіброзу. TGF-β1 додатково стимулює гіпертрофію ниркових клітин і накопичення ECM, служачи ключовим каналом, що призводить до тубулоінтерстиціального фіброзу. У нирковій інтерстиції конститутивно експресується рецептор фактора росту тромбоцитів-альфа (PDGFr-α). Експресія його ліганду, PDGF-CC, індукується через інфільтрацію моноцитів і макрофагів. Цей ліганд, своєю чергою, запускає каскад сигнальних шляхів, що охоплює Янус-кіназу/сигнальні перетворювачі та активатори транскрипції (JAK/STAT), PI3K, фосфоліпазу C-γ (PLC-γ) і MAPK, остаточно регулюючи експресію генів. Ці складні шляхи посилюють проліферацію фібробластів, міграцію та синтез ECM. Експерименти на мишах підтвердили, що переважна експресія PDGF-¬DD в інтерстиціальних фібробластах є очевидною як на ранніх, так і на пізніх стадіях запалення нирок. Крім того, PDGF-CC у перитубулярному капілярному ендотелії здійснює свій профіброзний вплив шляхом безпосередньої індукції проліферації фібробластів і посилення лейкоцитарної інфільтрації, що разом призводить до розвитку тубулоінтерстиціального фіброзу. Крім того, рівні HG стимулюють прозапальну, профіброзну та ангіогенну передачу сигналів у канальцях. Це спонукає до вироблення IL-6, CCL-2 і TGF-β, причому частина цієї відповіді опосередковується брадикініном (BK), опосередкованим сигнальним шляхом MAPK p42/p44.
Покращені кінцеві продукти глікації
AGE утворюються в результаті неферментативних реакцій між відновлюючими цукрами та білками, амінокислотами та іншими молекулами. Примітно, що проксимальний каналець служить основним місцем для реабсорбції AGE. Присутність AGE викликає експресію інтерлейкіну-8 (IL-8) і розчинного ICAM-1 у PTEC. Це, своєю чергою, сприяє інфільтрації запальних клітин у тубулоінтерстиціальний простір. AGE також здатні активувати Т-клітини CD4+ і CD8+, які проникають у мезенхіму та секретують цитокіни, такі як інтерферон-гамма (IFN-γ) і TNF-α. Ця запальна реакція, що супроводжується окислювальним стресом, сприяє запаленню тканин у нирковому середовищі. Отже, це запальне середовище призводить до порушення функції бар’єру капілярної стінки клубочка та підвищення проникності альбуміну. Крім того, AGE ініціюють активацію шляху PKC, згодом індукуючи експресію медіаторів запалення, включаючи ICAM-1, молекули адгезії судинних клітин (VCAM-1) і MCP-1. Це спостереження передбачає ключову роль PKC у контексті DKD.
Рекомендуємо переглянути запис семінару «Артеріальні гіпертензії ендокринного генезу» від провідного спеціаліста, кандидата медичних наук Костянтина Зуєва!
У контексті цукрового діабету збільшується кількість сайтів зв’язування AGEs у проксимальних ниркових канальцях. Зв’язуючись зі своїми рецепторами, AGE можуть індукувати вироблення TGF-β1. Після зв’язування з рецептором активний TGF-β1 переміщується в ядро шляхом рекрутування та фосфорилювання Smad, зокрема Smad2 і Smad3, тим самим регулюючи експресію генів, пов’язаних із виробництвом колагену. Кістковий морфогенетичний протеїн-7 (BMP-7) служить природним антагоністом TGF-β і має здатність протидіяти нирковому тубулоінтерстиціальному фіброзу, залучаючи шлях Smad1/5. Це передбачає утворення комплексів із Smad4, ядерну транслокацію та фосфорилювання Smad3. Крім того, варто зазначити, що Smad-незалежні сигнальні шляхи, пов’язані з TGF-β, також залучені, включаючи активацію p38 MAPK, c-Jun N-кінцевої кінази (JNK) і Rho. AGE стимулюють експресію фактора росту сполучної тканини (CTGF) в епітеліальних клітинах ниркових канальців. Сигнальний шлях ERK/p38-Smad3 взаємодіє з сигнальною віссю рецепторів епідермального фактора росту (ERGF)/p38-Smad3, суттєво сприяючи розвитку тубулоінтерстиціального фіброзу нирок. Важливо, що цей механізм працює незалежно від канонічного сигнального шляху TGF-β.
Метаболізм жирних кислот
Епітеліальні клітини канальців віддають перевагу виробленню енергії шляхом окислення жирних кислот (FAO) на відміну від метаболізму глюкози. Мітохондріальна окислювальна активність жирних кислот глибоко впливає на канальцеву реабсорбцію. Отже, метаболізм ліпідів і підтримка мітохондріальної функції проксимальних канальців мають величезне значення в клітинах ниркових канальців. Шкідливий вплив ліпотоксичності прискорює зниження функції канальців, підкреслюючи критичну роль відкладення ліпідів у ниркових канальцях, що пояснюється наступним чином:
- CD36-опосередковане відкладення ліпідів: трансмембранний глікопротеїн CD36, відповідальний за довголанцюговий транспорт жирних кислот, значно впливає на накопичення ліпідів у тканинах. Рецептор γ, активований проліфератором пероксисом (PPAR γ), ліганд-активований фактор транскрипції, регулює експресію та функціональність CD36. Дослідження демонструють, що HG активує сигнальний шлях протеїнкінази B/(AKT)-PPARγ, що призводить до підвищення мРНК CD36 і білка в епітеліальних клітинах канальців. Ця підвищена експресія CD36 посилює поглинання клітинами вільних жирних кислот, що в кінцевому рахунку сприяє зниженню життєздатності канальцевих клітин і відкладенню ліпідів. Націлювання на інгібування шляху AKT-PPARγ може мати терапевтичний потенціал для обмеження накопичення ліпідів у DKD.
- Nϵ-карбоксиметиллізин (CML) і RAGE-опосередковане накопичення ліпідів: CML, AGE, відіграє певну роль у ліпідному гомеостазі. ХМЛ зв’язується зі своїм рецептором RAGE, який експресується на поверхні епітеліальних клітин ниркових канальців. Ця взаємодія запускає внутрішньоклітинні сигнальні шляхи, які посилюють синтез холестерину через 3-гідрокси-3-метилглутарил-коензим А-редуктазу (HMG-CoAR) і підсилюють поглинання холестерину, опосередковане рецепторами ліпопротеїнів низької щільності (LDLr), через стрес ендоплазматичного ретикулуму (ERS). Одночасно ХМЛ зменшує відтік холестерину, опосередкований АТФ-зв’язуючим касетним транспортером A1 (ABCA1), спільно спричиняючи накопичення ліпідів у клітинах HK-2. Цей процес зрештою завершується утворенням трубчастих пінистих клітин.
- Кластерний сортувальний білок 2 (PACS-2) і накопичення ліпідів у канальцях: PACS-2, багатофункціональний сортувальний білок, який переважно експресується в ниркових канальцях, відіграє ключову роль у метаболізмі ліпідів. У дослідженнях за участю мишей із індукованим стрептозотоцином діабету та пацієнтів із DKD спостерігалося зниження рівня експресії PACS-2. Примітно, що специфічна для діабетичних канальців делеція Pacs-2 підсилює синтез ліпідів через посилену експресію стерол-О-ацилтрансферази 1 (SOAT1) і одночасне інгібування відтоку холестерину. Це посилене накопичення ліпідів у клітинах канальців сприяє прогресуванню екскреції альбумінурії та посилює ураження нирок.
Таким чином, накопичення внутрішньоклітинних ліпідів може ініціювати каскад подій, включаючи ERS, підвищене виробництво активних форм кисню (ROS) і стимулювання запальних реакцій. Феномен ліпотоксичності викликає мітохондріальну дисфункцію, спонукаючи епітеліальні клітини ниркових канальців приймати фіброзні фенотипи, що характеризуються підвищеним споживанням АТФ, загибеллю клітин, дедиференціацією та внутрішнім відкладенням ліпідів. Цей сукупний процес зрештою завершується розвитком тубулоінтерстиціального фіброзу.
Протеїнурія
За нормальних фізіологічних умов білки, які фільтруються з клубочка, зазнають майже повної реабсорбції в проксимальних канальцях. У контексті DKD поява мікроальбумінурії може бути пов’язана з порушенням здатності канальців реабсорбувати альбумін, а не в основному спричинена пошкодженням клубочків. Підвищена кількість відфільтрованих білків служить стимулом для сигнального шляху PTEC, що призводить до аномальних регуляторних реакцій, які охоплюють такі явища, як ріст тубулярних клітин, апоптоз, зміни в транскрипції генів і подальша індукція запальних факторів, які сприяють запаленню та фіброзу. Якщо розглядати з точки зору запалення, протеїнурія виступає як ключовий прозапальний стимул в активації NF- κB в клітинах ниркових канальців. Хемокіни та молекули адгезії, організовані через NF-κB-залежний шлях, також посилюються через надмірну кількість білків ультрафільтрації в проксимальних канальцевих клітинах. Таким чином, можна стверджувати, що активація NF-κB і транскрипція специфічних прозапальних хемокінів є ознаками прогресуючої DKD. Конкретним прикладом є хемокін 1 мотиву C-X3-C (CX3CL1), пов’язаний з мембраною хемокін, який стимулює регуляцію CX3CR1 у проксимальних канальцевих епітеліальних клітинах через NFκB та p38–MAPK-залежні шляхи. Подібним чином підвищена експресія хемокіну 5 мотиву CC (CCL5, також відомого як RANTES) очевидна у зразках біопсії нирок пацієнтів із цукровим діабетом 2 типу, що переважно спостерігається в клітинах ниркових канальців. Перевантаження білками викликає регуляцію CCL5, і ступінь експресії CCL5 у канальцевих клітинах безпосередньо пов’язаний з об’ємом протеїнурії та інфільтрацією інтерстиціальних клітин. Комбіновані ефекти CX3CR1 та CCL5 сприяють інтерстиціальному запаленню та прогресуванню захворювання. сприяючи рекрутуванню та адгезії моноцитів, Т-клітин і природних клітин-кілерів у перитубулярному інтерстиції. Слід зазначити, що дослідження Галкіної та ін. демонструє, що основним місцем рекрутування Т-клітин є строма, особливо підкреслюючи значне збільшення (у 6-10 разів) клітин CD4+, CD8+ і CD20+ в інтерстиції. Крім того, велика кількість клітин CD4+ і CD20+ корелює зі ступенем протеїнурії. Інфільтрація інтерстиціальних Т-клітин суттєво корелює зі ступенем протеїнурії, що вказує на імунопатологічний механізм, що лежить в основі, який сприяє прогресуванню протеїнурії та інтерстиціального запалення при DKD. Рівні VCAM-1 відповідають кількості імунних клітин, що проникають у нирки, і вони також пов’язані з тяжкістю та прогресуванням протеїнурії. Крім того, білок сечі може ініціювати пошкодження канальцевих клітин, активуючи систему комплементу та стимулюючи канальцеві клітини генерувати активні форми кисню.
Рекомендуємо переглянути запис семінару «Що потрібно знати про предіабет сімейному лікарю» від провідного експерта, професора Ольги Барни!
З точки зору сприяння розвитку фіброзу TGF-β в цей час розглядається як прозапальний і фіброзний медіатор, індукований впливом альбуміну. Коли білок присутній у сечі, він зв’язується зі своїми рецепторами, викликаючи вивільнення цитокінів, які ще більше сприяють запаленню та фіброзу. Цей кумулятивний ефект в кінцевому підсумку призводить до зниження функції нирок. Було виявлено, що наявність надлишку білка в сечі регулює експресію TGF-β і стимулює відкладення позаклітинного матриксу ECM, тим самим посилюючи фіброзні реакції. У мишей, індукованих стрептозотоцином (STZ) і з нокаутом протеїнкінази С-епсилон (PKC-ϵ), спостерігалося значне збільшення мікроальбумінурії, тубулоінтерстиціального фіброзу та мезангіальної дилатації. Це свідчить про те, що нирковий фіброз, опосередкований делецією PKC-ϵ, може включати сигнальний шлях TGF-β1. Інша форма PKC, PKC-β, демонструє підвищену активність у ниркових канальцях. Показано, що інгібування PKC-β зменшує накопичення ниркових макрофагів, експресію запальних молекул (таких як ICAM-1 і MCP-1) і тубулоінтерстиціальне пошкодження. Під час пошкодження ниркового фіброзу Klotho відіграє певну роль у інгібуванні TGF-β1-індукованої реакції епітеліально-мезенхімального переходу (EMT) у культивованих клітинах. Ця дія характеризується зниженням експресії епітеліальних маркерів, інтерстиціальних маркерів і міграції клітин. Експериментальні дослідження припустили, що цей ефект може бути наслідком того, що виділений Klotho безпосередньо зв’язується з рецепторами TGF-β ІІ типу, тим самим перешкоджаючи зв’язуванню TGF-β1 з рецепторами клітинної поверхні та, як наслідок, пригнічуючи передачу сигналу TGF-β1. Крім того, було виявлено, що секретований Klotho перешкоджає відповіді EMT шляхом інгібування сигнальних шляхів Wnt та IGF-1. Рівні сироваткового розчинного Klotho (sKlotho) демонструють значну негативну кореляцію з різним ступенем альбуміну в сечі у пацієнтів із цукровим діабетом 2 типу (ЦД 2). Це розуміння свідчить про те, що sKlotho може служити потенційним біомаркером для прогнозування прогресування захворювання нирок у майбутньому.
Надлишок білка в сечі може призвести до посилення експресії генів у клітинах ниркових канальців, що призводить до надмірної експресії різних хемокінів. Це, своєю чергою, спричиняє агрегацію імунних клітин, таких як моноцити та Т-клітини, усередині тубулоінтерстицію. Вивільнення IL додатково приваблює нейтрофіли до агрегації, і це запальне середовище сприяє синтезу волокнистих молекул, таких як ангіотензин II (Ang II) і TGF-β. Ці процеси разом сприяють деградації базальної мембрани канальців, сприяючи проникненню запальних клітин в інтерстицій і капілярний простір, що оточує ниркові канальці. Зрештою, ця послідовність подій викликає появу фіброзу. Таким чином, надмірна кількість білка в сечі стимулює епітеліальні клітини проксимальних канальців, встановлюючи зв’язок між протеїнурією, інтерстиціальним запаленням і фіброзом.
Окислювальний стрес
Стійка гіперглікемія призводить до збільшення AGEs, поступового посилення активності Na+-K+-АТФази в канальцях і аномального метаболізму епітеліальних клітин. Ці фактори сприяють мітохондріальній дисфункції та виробленню великої кількості АФК. Отже, ендотеліальні клітини зазнають пошкодження, що призводить до залучення запальних клітин і факторів запалення до місця пошкодження. Цей процес запускає тубулоінтерстиціальну запальну відповідь. У контексті досліджень in vivo було виявлено, що надмірна експресія Klotho ефективно знижує експресію ROS. Це зниження відбувається через послаблення стимуляції TNF-α в нирках. У результаті надмірна експресія Klotho пригнічує активацію NF-kB і, отже, пригнічує продукцію запальних цитокінів, хемокінів і факторів росту. Ця дія ефективно пригнічує реакцію на окислювальний стрес. Однак важливо відзначити, що експресія Klotho зазвичай низька у випадках DKD. Останні дослідження підкреслюють, що CD36 є потенційним ключовим медіатором продукції АФК при хронічній хворобі нирок. Блокування CD36 перериває ЕМТ, індукований високим вмістом глюкози, у клітинах епітелію канальців. Це переривання в основному відбувається через сигнальні шляхи ERK1/2 і TGF-β1/Smad2.
Діабетичне середовище значно сприяє виробленню різних хемокінів, включаючи MCP-1, OPN, CCL-2, CX3CL1, INF-γ-індукований білок (CXCL10) і CCL5. Ці хемокіни відіграють вирішальну роль у залученні запальних клітин (макрофагів і Т-клітин) до ниркових канальців та інтерстицію, таким чином встановлюючи запальний цикл. Інфільтрація клітин запалення в канальцях може призвести до розривів і потовщення базальної мембрани канальців, що зрештою призводить до атрофії канальців на пізніх стадіях. Інфільтрація запальних клітин в нирковий інтерстицій призводить до вивільнення прозапальних, профіброзних і антиангіогенних факторів, що в кінцевому підсумку викликає інтерстиціальний фіброз (IFTA). Цікаво, що результати біопсії нирок виявляють наявність агрегатів інтерстиціальних еозинофілів (ІЕА) у пацієнтів із ДЗН. Ступінь тяжкості IEA корелює з IFTA, що ускладнює остаточну діагностику алергічного інтерстиціального нефриту. Це вказує на те, що МЕА при DKD може або відображати запальну відповідь на хронічне тубулоінтерстиціальне пошкодження, або потенційно служити стимулом, що сприяє такому пошкодженню, або навіть комбінацією обох факторів. Опублікована стаття в першу чергу наголошує на тубулоінтерстиціальному запаленні, тоді як для повного розуміння тубуліту потрібні подальші спостереження та дослідження.
Механізм ішемії та гіпоксії
Кровопостачання епітеліальних клітин канальців в основному відбувається з еферентних артеріол клубочків, зрештою досягаючи вен, що оточують ниркові канальці. Цей процес складно регулюється різними нейрогуморальними факторами. Пошкодження, викликане гіпоксією проксимальних канальців при DKD, можна пояснити трьома основними факторами:
- Зниження кровотоку: Зниження кровотоку в перитубулярних капілярах (ПТК) в основному виникає внаслідок дисрегуляції факторів вазоконстрикції та релаксації. У присутності AGE та підвищених рівнів глюкози фосфорилювання та експресія ендотеліальної синтази оксиду азоту (eNOS) знижуються, що призводить до дисфункції та пошкодження ендотеліальних клітин судин. NO, вазоактивна речовина, відіграє вирішальну роль у сприянні вазодилатації та захисті ендотеліальних клітин судин. Однак його синтез знижений. Одночасно Ang II сприяє звуженню як аферентних, так і еферентних артеріол. Він сприяє проліферації ендотеліальних клітин, гіпертрофії та зменшує кровопостачання перитубулярних капілярів.
- Порушення утилізації кисню: процес транспорту натрій-глюкози через мембрану клітин проксимальних канальців не є енергоємним, але залежить від активності Na+/K+ АТФази. У діабетичних щурів на ранній стадії захворювання спостерігалося порушення виробництва АТФ у мітохондріях і фрагментація органоїдів у проксимальних епітеліальних клітинах канальців. Це пов’язано з підвищеною екскрецією альбуміну з сечею, аномальною морфологією клубочків і навіть підвищеним рівнем молекули ураження нирок сечі-1 (KIM-1). Ці мітохондріальні структурні та функціональні аномалії можуть являти собою найбільш ранні прояви DKD. У пацієнтів із ДЗЗ посилюється реабсорбція глюкози, підвищується метаболічна активність, підвищується споживання О2, а структура та дисфункція мітохондрій призводять до порушення утилізації кисню. У результаті проксимальний каналець стає більш сприйнятливим до ішемічного ураження та має тенденцію до гострого пошкодження нирок (ГПН).
- Розрідження мікросудин: враховуючи погану толерантність ниркової тканини до гіпоксії та життєво важливу реабсорбційну функцію ниркових канальців, розвиток складної та щільної мікросудинної мережі є важливим. У контексті DKD існує дисбаланс між проангіогенними та антиангіогенними факторами. Дослідження на тваринах показують, що рівні мРНК і білка VEGF знижуються, в той час, як експресія тромбоспондину-1 (TSP-1), інгібітора ангіогенезу, збільшується. Ці зміни призводять до набряку ендотелію, втрати капілярів і протеїнурії. Гіпоксія сприяє збільшенню виробництва позаклітинного матриксу через як TGF-β-залежні, так і незалежні механізми. Гіпоксія-індукований фактор (HIF) підвищує експресію CTGF. Це призводить до збільшення відстані дифузії для доставки кисню до паренхіми. Фіброзне розширення інтерстицію ще більше стискає та порушує локальну мікросудинну мережу, зрештою викликаючи мікросудинне розрідження. Це посилює ступінь тубулоінтерстиціальної гіпоксії, встановлюючи шкідливу петлю зворотного зв'язку.
Гіпоксія є вирішальним фактором, що лежить в основі виникнення та прогресування DKD. Гемодинамічні зрушення, метаболічні впливи та імунні тригери можуть безпосередньо завдати шкоди судинним ендотеліальним клітинам, що призводить до місцевої активації RAS або зниження рівня NO. Це викликає звуження ниркових судин і перешкоджає доставці кисню. Оскільки транспорт кисню зменшується, це порушує ниркову перфузію, посилюючи ниркову гіпоксію. Як наслідок, епітеліальні клітини ниркових канальців відчувають мітохондріальну дисфункцію та порушення утилізації кисню, що призводить до клітинної дегенерації, атрофії, пошкодження перидуктальних капілярів і подальшого зниження кровопостачання. Ці каскадні ефекти сприяють розвитку інтерстиціального фіброзу та погіршенню функції нирок.
Шкідливі біомаркери для епітеліальних клітин ниркових канальців
Молекула пошкодження нирок-1
KIM-1 є глікопротеїном, що експресується в проксимальних канальцях і служить чутливим індикатором пошкодження канальців. Хоча він відсутній у нормальних нирках, його експресія значно зростає під час гострого пошкодження або запалення нирок. KIM-1, виявлений на PTEC, відіграє роль фагоцитарної клітини, очищаючи клітинне сміття та апоптотичні тіла всередині пошкодженого тубулоінтерстиціального відділу. Ця дія сприяє регенерації пошкоджених канальців. Однак KIM-1 також полегшує поглинання жирних кислот тубулярними клітинами, сприяючи прогресуванню прогресуючої DKD. Наявність KIM-1 корелює з тубулоінтерстиціальним запаленням і фіброзом. У пацієнтів із DKD KIM-1 тісно пов’язаний із ризиком прогресуючого зниження функції нирок, і його підвищення спостерігалося у підтверджених випадках DKD. KIM-1 є відносно нестабільним, його позаклітинний домен розщеплюється, потрапляє в просвіт канальців і в кінцевому підсумку виводиться з сечею. При DKD рівень KIM-1 у сечі свідчить про раннє ураження канальців. Зараз KIM-1 в сечі широко визнаний як специфічний і чутливий біомаркер для оцінки ураження проксимальних канальців нирок.
Ліпокалін, асоційований з нейтрофільною желатиназою
NGAL є глікопротеїном 25 кДа, який широко поширений у клітинах тканин людини. Його експресія в нирковій тканині мінімальна за нормальних умов. NGAL переважно експресується в петлях Генле та дистальних канальцях, відіграючи роль у метаболізмі заліза шляхом зв’язування з транспортерами заліза. NGAL сприяє регенерації канальцевих епітеліальних клітин, індукуючи апоптоз інфільтрованих нейтрофілів усередині тубулоінтерстицію, тим самим захищаючи ниркову тканину від запального пошкодження клітин. У випадках ішемії та гіпоксії нирок експресія NGAL у клітинах епітелію канальців значно посилюється. Пошкоджені тубулярні клітини виробляють NGAL, який згодом секретується в кров і сечу. Рівень NGAL у сечі може виявити AKI протягом 2 годин, тоді як значні зміни креатиніну в сироватці крові тривають від 3 до 4 днів, що підкреслює роль NGAL як раннього та більш чутливого індикатора AKI порівняно з креатиніном сироватки. Дослідження також продемонстрували сильну кореляцію між рівнями NGAL у сечі та атрофією канальців. У випадках запального ураження моноцити/макрофаги, нейтрофіли та клітини епітелію канальців є первинними джерелами продукції NGAL. Примітно, що пошкодження канальців може проявлятися раніше, ніж пошкодження клубочків, при запальних станах DKD. Як маркер раннього ушкодження канальців, NGAL може виявити порушення функції нирок у хворих на діабет до появи мікроальбуміну в сечі (mALB).
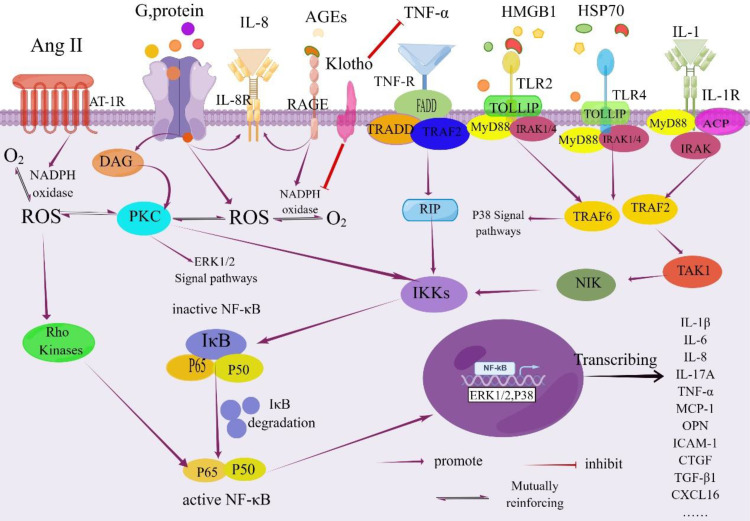
Таким чином, гіперглікемія служить ініціюючим фактором для тубулоінтерстиціального пошкодження при ДЗН, запускаючи каскад клітинних відповідей. Це включає накопичення AGEs, активацію окислювального стресу, підвищення внутрішньониркових рівнів ангіотензину II та посилення експресії прозапальних і профіброзних факторів. Ці процеси разом сприяють атрофії канальців, інтерстиціальному запаленню та фіброзу, які є ключовими факторами прогресування DKD.
Тубулоінтерстиціальний фіброз тісно переплітається з наявністю інфільтруючих запальних клітин. У відповідь на ці клітини, сигнали стресу та різні медіатори, зокрема TGF-β, канальцеві епітеліальні клітини зазнають трансформації в мезенхімальні клітини, процес, відомий як епітеліально-мезенхімальний перехід. Експресію TGF-β регулюють кілька факторів, у тому числі надлишок альбуміну, гіперглікемія, стимуляція ангіотензином II і активація сигнальних шляхів, таких як MAPK, JAK-STAT і PKC. Крім того, Smad-незалежний сигнальний шлях TGF-β, який може включати активацію p38 MAPK, JNK і Rho, також відіграє певну роль. Ці складні сигнальні шляхи спільно впливають на експресію генів, зрештою посилюючи проліферацію, міграцію та синтез фібробластів ECM.
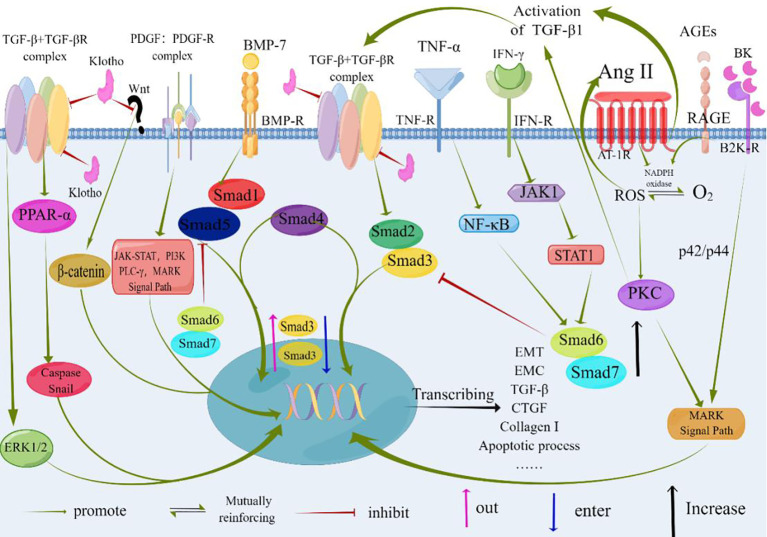
Терапевтичні мішені при тубулоінтерстиціальних ураженнях нирок
Після схвалення першого інгібітора АПФ, каптоприлу, FDA США в 1981 році для зменшення протеїнурії та уповільнення прогресування функції нирок, дослідження нових блокаторів РАН стало фундаментальним аспектом лікування різних захворювань нирок. Початкові дослідження в першу чергу були зосереджені на перевагах блокаторів РАН щодо захисту нирок через посилення регуляції системного артеріального тиску та корекцію аномальної ниркової гемодинаміки. З усім тим, починаючи з 2001 року, дослідження виявили, що система RAS також демонструє відмінні протизапальні ефекти та ефекти, що пригнічують прогресування ХХН, які не пов’язані лише з її здатністю знижувати кров’яний тиск. В останні роки, як розуміння Основні механізми DKD поглибилися, і з’явилися значні результати великомасштабних рандомізованих контрольованих досліджень (РКД), інгібітори SGLT-2 і нестероїдні антагоністи мінералокортикоїдних рецепторів стали ефективними терапевтичними варіантами для лікування DKD. Ці досягнення знайшли широке впровадження в клінічну практику.
Інгібітори SGLT-2
Захисна дія SGLT-2i на нирки включає як гемодинамічні, так і негемодинамічні механізми. З гемодинамічної точки зору SGLT-2i пригнічує функцію SGLT-2 у проксимальних канальцях нирок, зменшуючи реабсорбцію натрію та глюкози. Це призводить до збільшення концентрації натрію та глюкози в товстій висхідній кінцівці петлі Генле, спонукаючи юкстагломерулярні клітини вивільняти ренін. Цей процес посилює тубулогломерулярний зворотний зв'язок і індукує звуження приносних артеріол. Таким чином, покращується стан високої ниркової перфузії, тиску та фільтрації в клубочках, що сприяє покращенню функції нирок. Цей механізм має ключове значення для полегшення розвитку та прогресування DKD. Крім того, SGLT-2i пом’якшує тубулоінтерстиціальне пошкодження при DKD, надаючи протизапальну та антиоксидантну дію за допомогою таких механізмів:
- Регуляція ангіотензину II: Ang II посилює експресію SGLT1 і SGLT2 в епітеліальних клітинах ниркових канальців, що призводить до збільшення поглинання глюкози та підвищення внутрішньоклітинних рівнів ROS, викликаючи окислювальний стрес. SGLT2i ефективно протидіє цьому підвищеному поглинанню глюкози.
- Ендотеліальна функція: Ang II знижує регуляцію функції eNOS, що призводить до ендотеліальної дисфункції та протромботичного стану. Зменшення синтезу оксиду азоту призводить до збільшення виробництва молекул адгезії, тканинного фактора, MCP-1 і активності β-галактозидази. SGLT2i полегшує фосфорилювання AMPK, відновлює нормальну аутофагію, знижує активність рапаміцину (mTOR) у ссавців і зменшує утворення факторів запалення, таких як інтерлейкін 1β (IL-1β), IL-6 та TNF-α. Крім того, пентраксин-3 (PTX3), протеїн, який стимулює фенотип M2 макрофагів, який знижує регуляцію NF-κB, IL-1β, TNF-α та MCP-1, часто значно знижується у пацієнтів з ДЗН.
- Інгібування епітеліально-мезенхімального переходу (EMT): при прозапальній стимуляції та надмірній протеїнурії тубулярні епітеліальні клітини зазнають переходу в міофібробласти. HG стимулює синтез і вивільнення матриксної металопротеїнази 2 (MMP2), яка руйнує базальну мембрану канальців, спричиняючи фенотипічно змінені клітини епітелію канальців від’єднуватися та мігрувати до інтерстицію. Ці клітини разом із резидентними нирковими фібробластами згодом виробляють надмірну кількість протеїнів позаклітинного матриксу, що призводить до інтерстиціального фіброзу. SGLT2i перешкоджає диференціації ендотеліальних клітин-попередників (EPC) або клітин з характеристиками EPC в міофібробласти, таким чином помітно зменшуючи кількість клітин, які піддаються ЕМТ всередині канальців. Крім того, експресія HIF-1α, викликана гіпоксією, сприяє ЕМТ та інтерстиціальному фіброзу шляхом активації TGF-β-залежного шляху та сигнального шляху PI3K/Akt. Таким чином, підвищення рівня гемоглобіну у пацієнтів з DKD також може означати відновлення після тубулоінтерстиціального пошкодження та фіброзу, викликаного HG (152, 153). SGLT2i ефективно зменшує генерацію мікроРНК-21, індуковану високим рівнем глюкози, яка спрямована на RECK — білок, що містить мотив Kazal. який пригнічує матриксну металопротеїназу 2, таким чином пригнічуючи ЕМТ та міграцію клітин. Це ще один потенційний механізм захисту нирок.
Останні РКД, включаючи CREDENCE, DAPA-CKD та EMPA-KIDNEY, надали переконливі докази того, що комбінація інгібіторів SGLT2 та інгібіторів РАН у осіб із цукровим діабетом 2 типу призводить до зниження ризику ниркової та серцево-судинної кінцевої точки. Однак, ці дослідження в першу чергу наголошують на клінічних результатах лікування та прогнозах, не маючи повного уявлення про прогресування канальцевих та інтерстиціальних уражень.
Рекомендуємо переглянути запис семінару «Предіабет: лікувати чи ні?» від провідного спікера, професора Юлії Комісаренко!
Агоністи глюкагоноподібного пептиду-1
Крім зниження рівня глюкози, агоністи GLP-1 виявляють захисну дію на нирки, включаючи протизапальні, антиоксидантні властивості та зниження протеїнурії. Дослідження на тваринних моделях показують, що агоністи GLP-1 можуть зменшувати протеїнурію та інфільтрацію макрофагів у мишей, одночасно знижуючи рівні ICAM-1, TGF-β і колагену типу IV. Механізм, що лежить в основі цих ефектів, потенційно передбачає стримування вироблення запальних цитокінів (IL-1β, TNF-α) через шляхи впливу Ang II і AGE. Ця дія призводить до інгібування NF-κB, активації Rho-кінази та сигнальних шляхів, таких як PKC і PKA, в кінцевому підсумку стримуючи запалення та окислювальний стрес.
У клінічних дослідженнях застосування ексенатиду або ліраглутиду продемонструвало ефективний захист передбачуваної ШКФ у пацієнтів з цукровим діабетом, а також зменшило мікроальбумінурію та зменшило виділення з сечею TGF-β1 і колагену типу IV. Цей нирковий захисний ефект, здається, не залежить від контролю глікемії.
Антагоністи мінералокортикоїдних рецепторів
Спочатку було виявлено, що альдостерон діє на епітеліальну тканину дистальних канальців у нирках. Ця дія, опосередкована мінералокортикоїдним рецептором (MR), полегшує реабсорбцію натрію та секрецію калію, зрештою сприяючи підвищенню артеріального тиску шляхом розширення позаклітинного об’єму. Однак надмірна активність MR-рецепторів не тільки сприяє реабсорбції натрію та води, що призводить до збільшення об’єму крові, але також викликає утворення АФК і прозапальних факторів. Ці процеси сприяють фіброзу нирок, запальним реакціям і збільшенню нирок. Виникнення вивільнення альдостерону після застосування інгібіторів АПФ або БРА спонукало до застосування антагоністів альдостерону, таких як спіронолактон і еплеренон, у контексті лікування DKD.
На рівні цитоплазматичної мембрани стероїдні MRA мають хімічну структуру, подібну до альдостерону, і конкурують за його сайти зв’язування, таким чином надаючи прямі антагоністичні ефекти. Ця дія призводить до зменшення окислювального стресу, запалення та експресії профіброзних факторів. В результаті запальна відповідь і прогресування фіброзу гальмуються. Щоб подолати недостатню селективність спіронолактону та нижчу спорідненість еплеренону до МР, було розроблено нове покоління нестероїдних МРА, таких як фанеренон. Відмінні клітинні механізми Finerenone дають такі переваги, як менша частота пов’язаних з лікуванням гіперкаліємії та AKI, а також більш м’який вплив на систолічний артеріальний тиск. Фінеренон демонструє підвищену селективність і рецепторну активність порівняно з антагоністами мінералокортикоїдних рецепторів першого та другого покоління. Його переваги випливають з його відмінної молекулярної структури та механізму дії:
- Диференціація хімічної структури: розробка Finerenone базується на хімічній структурі блокатора дигідропіридинових каналів, але не має активності на кальцієві канали L-типу. Ця характеристика надає йому більшу полярність, ніж стероїдні MRA, і дозволяє йому проникати через гематоенцефалічний бар’єр, що має вирішальне значення для регуляції артеріального тиску.
- Значні відмінності в структурі розподілу в тканинах. Фінеренон демонструє збалансований розподіл у тканинах серця та нирок. Навпаки, стероїдні MRA, такі як спіронолактон і еплеренон, накопичуються в нирках на рівнях щонайменше в три рази вище. Отже, фінренон ефективніше знижує гіпертрофію міокарда, рівні промозкового натрійуретичного пептиду в плазмі та протеїнурію.
- Різні способи зв’язування з молекулярним рецептором: зі значним замісником файнренон утворює нестабільні комплекси MR-ліганд після зв’язування з MR. Ці комплекси не в змозі рекрутувати кофактори, що відрізняє їх спосіб зв’язування від інших MRA.
Фінеренон завдяки своїй дії, що блокує МР, сприяє покращенню структури та функції епітеліальних клітин ниркових канальців. Це покращення пов’язане зі зменшенням фосфорилювання S6K1 і зниженням окисного стресу. Основним ефектом є пригнічення генерації супероксид-аніонів, викликаного Ang II, PDGF та EGF (малюнок 4). Ця дія сприяє відновленню молекул адгезії та зменшує структурні пошкодження в проксимальних тубулярних клітинах (PTC). На ранніх стадіях інтерстиціального фіброзу втрата комплексу молекула адгезії-ламінін сприяє порушенню адгезії ПТК. Антагоністичний вплив МР покращує комплекси фіброз-ламінін-кальцій-зв'язуючий білок шляхом відновлення спайок. Примітно, що ефекти МР-системи залежать від таких факторів, як ліганд (альдостерон), клітинне середовище та промотори цільових генів. MR у поєднанні зі своїми факторами спільної регуляції функціонує спільно на цих шляхах. Фінеренон ефективно затримує утворення комплексів MR-альдостерон і перешкоджає рекрутуванню важливих кофакторів транскрипції. Крім того, він успішно пригнічує експресію тенасцину-X (визнаного профіброзного фактора), підвищує біодоступність оксиду азоту, знижує рівень ROS, пом’якшує ендотеліальну дисфункцію, гіпертрофію міокарда та протеїнурію, а також підтримує протизапальну та антифіброзну дію.

Нещодавнє дослідження, опубліковане в JCI Insight, показало, що антагоністи МР мають здатність зменшувати екскрецію білка з сечею та перешкоджати прогресуванню DKD, захищаючи гломерулярний ендотеліальний глікокалікс (GEnGlx). GEnGlx являє собою гідратований поліаніонний гель, який регулює проникність судин і полегшує механічну передачу напруги зсуву. Цей глікокалікс є невід’ємною частиною бар'єра клубочкової фільтрації, і MRAs знижують активність матриксної металопротеїнази (ММР), зменшуючи пошкодження GEnGlx. Отже, це зменшує проникність клубочків і протеїнурію в осіб з діабетом.
З 2013 року клінічні випробування з участю фінренону, такі як Дослідження переносимості антагоністів рецепторів мінералокортикоїдів (ARTS), оприлюднили багатообіцяючі клінічні результати. Ці відкриття ознаменували нову еру в лікуванні DKD. Слід зазначити, що нещодавно опубліковані дослідження FIDELIO-DKD і FIGARO-DKD є додатковими дослідженнями, які оцінюють переваги фінренону для нирок і серця у пацієнтів з DKD. Фінеренон не тільки суттєво послаблює зниження eGFR і покращує альбумінурію, але й суттєво знижує ризик прогресування DKD. Крім того, сполука значно зменшує частоту нової серцевої недостатності та знижує ризик серцево-судинної смерті та нефатального інфаркту міокарда. Схвалення фінеренону знаменує початок ери «потрійної терапії» у лікуванні DKD.
Потенційні терапевтичні мішені для лікування DKD
Високий рівень глюкози в крові сприяє вивільненню HMGB1, ендогенного ліганду TLR і рецептора RAGE, з тубулярних епітеліальних клітин і клубочкових подоцитів. HMGB1 складається з блоку A та блоку B, причому блок B зв’язується з TLR2, TLR4 та RAGE, активуючи сигнальний шлях NF-κB та індукуючи прозапальні реакції. І навпаки, блок А може пригнічувати вироблення прозапальних цитокінів, стимульованих HMGB1. Зараз терапевтичні стратегії, спрямовані на HMGB1, здебільшого зосереджені навколо розчинних рецепторів для кінцевих продуктів прогресуючого глікування (esRAGE) і рекомбінантного блоку HMGB1 A. esRAGE, сплайс-варіант гена RAGE, може секвеструвати позаклітинні ліганди RAGE, пригнічуючи активацію RAGE на поверхні клітини. Він перешкоджає зв'язуванню HMGB1 з RAGE, TLR2 і TLR4. Клінічне дослідження продемонструвало, що надмірна експресія esRAGE може зменшити протеїнурію, захворювання клубочків і діабетичне ураження нирок. Крім того, TLR4 має тісний зв’язок із тубулоінтерстиціальним запаленням при DKD. Посилена активність TLR4 запускає сигнальний шлях NF-κB і викликає запальні реакції. TLR4 бере участь у опосередкуванні тубулоінтерстиціального запалення при DKD. Отже, інгібування шляху TLR4/NF-κB може запропонувати новий терапевтичний шлях для DKD. TLR2 і TLR4 сильно експресуються в ниркових канальцях, і їхні рівні були пов’язані з інтерстиціальною макрофагальною інфільтрацією, рівнями HbA1c і eGFR під час біопсії нирки. Таким чином, націлювання на HMGB1 за допомогою рекомбінантного блоку A або інгібування/блокування TLR може потенційно пом’якшити протеїнурію, пошкодження клубочків, інтерстиціальний фіброз і запалення нирок при DKD, пропонуючи проспективний терапевтичний підхід.
Протеїн Klotho, однопрохідний трансмембранний білок, який в основному міститься в епітеліальних клітинах ниркових канальців, забезпечує захист нирок, надаючи антистарільний, протизапальний, антиоксидантний і антифіброзний ефекти (4, 105). Клінічні дані дослідження PREDIAN підкреслюють, що комбінація спіронолактону з блокаторами РАН запобігає зниженню рівня білка Klotho. Крім того, це комбіноване лікування пригнічує експресію TNF-α та TNF-подібного слабкого індуктора апоптозу (TWEAK) у епітеліальних клітинах ниркових канальців як додаткова терапія. Примітно, що сироваткові рівні розчинного Klotho (sKlotho) і NGAL демонструють значну негативну кореляцію у пацієнтів з DKD з різним ступенем альбумінурії. Ці маркери потенційно можуть служити прогностичними біомаркерами для прогресування ДЗЗ у майбутньому.
Фосфорилювання OPN запускає інфільтрацію макрофагів у нирковий інтерстицій. OPN помітно експресується в різних сегментах ниркових канальців, включаючи проксимальні канальці, дистальні канальці, товсті висхідні кінцівки медулярної частини та деякі епітеліальні клітини збиральних проток. Як функціональний неколагеновий білок, OPN відіграє роль у клітинній адгезії та рекрутуванні. Він індукує хемотаксис макрофагів, модулює сигнальний шлях NO та опосередковує вивільнення запальних і фіброзних факторів (таких як Ang II, TGF-β, ендотелін-1), які беруть участь у патогенезі DKD, сприяючи тубулоінтерстиціальному пошкодженню. Порівняно з MCP-1, OPN демонструє більш тісний патологічний зв’язок із ураженнями канальців, що стає важливою терапевтичною мішенню для лікування тубулоінтерстиціального запалення при ДЗН.
Сигнальний шлях NF-κB має ключове значення в розвитку запалення канальців. Дослідження зосереджено на інгібуванні активності NF-κB і регулюванні прозапальних цитокінів. Пов’язані препарати включають BAY 11-7082, інгібітор NF-κB PDTC, інгібітори кінази Rho та триптолід. Інгібування наступних прозапальних факторів NF-κB зменшує генерацію АФК, інфільтрацію макрофагів і пов’язану експресію запального фактора в нирках, що згодом пом’якшує ниркове інтерстиціальне пошкодження. Наприклад, пов’язаний з TNF ліганд, що індукує апоптоз (TRAIL), викликає апоптоз епітеліальних клітин ниркових канальців за високих рівнів глюкози та прозапального впливу цитокінів, що корелює зі ступенем тубулярної атрофії, інтерстиціального фіброзу та запалення. CCL5 помітно підвищується в епітеліальних клітинах ниркових канальців і прямо корелює з протеїнурією та інфільтрацією інтерстиціальних клітин. CXCL16, прозапальний цитокін, підвищується в епітеліальних та інтерстиціальних клітинах ниркових канальців при DKD. Активуючи шлях TLR4/NF-κB, CXCL16 загострює DKD, індукуючи запальну інфільтрацію клітин і посилений апоптоз клітин. Нещодавні дослідження підкреслили роль IL-17A у сприянні запаленню нирок і прогресуванню DKD. Інтерлейкін-17A (IL-17A), імунозалежний цитокін, стимулює запальні та імунні реакції. Зв’язуючись зі своїм рецептором IL-17RA, IL-17A запускає активацію тубулоінтерстиціальних клітин, викликає запальну клітинну інфільтрацію та викликає пошкодження канальців. Таким чином, інгібування шляхів CXCL16 та IL-17A/IL-17RA потенційно відкриває нову терапевтичну стратегію для DKD.
Рекомендуємо переглянути запис семінару «Відновлення судомоторної функції в пацієнта з ЦД 1 типу на прикладі клінічного випадку» від спікера Надії Жердьової!
TGF-α1, β2 і β3 є найпотужнішими промоторами накопичення ECM, причому шлях Smad є ключовим у передачі сигналу TGF-β в межах DKD. Цей шлях відіграє вирішальну роль у накопиченні внутрішніх ниркових клітин і молекул ECM. Придушення ефектів, опосередкованих TGF-β, і фосфорилювання Smad2 може полегшити нирковий інтерстиціальний фіброз, атрофію канальців і покращити інфільтрацію клітин запалення та проліферацію фібробластів у мишей. BMP-7, природний антагоніст TGF-β, демонструє потужні захисні функції нирок, ефективно усуваючи нирковий фіброз на різних тваринних моделях. Однак клінічні випробування BMP-7 для лікування захворювань нирок на сьогодні не повідомляються.
Крім того, сфера традиційної китайської медицини (ТКМ) і фітотерапії виявила протизапальну дію. Пеоніфлорин, наприклад, демонструє здатність зменшувати інфільтрацію макрофагів і експресію запальних цитокінів, посилюючи функцію нирок і полегшуючи гістологічне пошкодження в нирках діабетичних мишей. Глікозиди триптеригіуму виявляють протизапальні властивості та ефективно запобігають розриву клубочкової мембрани, спричиненого окисненням, що, як наслідок, зменшує протеїнурію при гломерулонефриті та перешкоджає прогресуванню DKD. Хоча докази низької якості свідчать про те, що триптеригіум може бути потенційною додатковою терапією для лікування DKD, більшість цих досліджень залишаються обмеженими клітинним, молекулярним і тваринним рівнями, з відсутністю великомасштабних проспективних досліджень, спрямованих конкретно на канальці ураження та високоякісні патологічні докази, що підтверджують їх ефективність.
Ці потенційні терапевтичні цілі та шляхи передачі сигналів дають нові ідеї та шляхи лікування DKD. Майбутні дослідження повинні глибше вивчати терапевтичні ефекти та безпеку цих мішеней і шляхів.
Висновки
Підсумовуючи, дослідження тубулоінтерстиціального запалення при ДЗЗ привернуло значну увагу та значення. Розуміння основних механізмів і визначення потенційних терапевтичних цілей мають вирішальне значення для розробки ефективних стратегій лікування. Розпізнавання потенційних мішеней, включаючи білок TLR4, HMGB1, MCP-1, OPN, BMP-7 і Klotho, разом із ключовою роллю, яку відіграють сигнальні шляхи NF-κB і TGF-β/Smad, представляє багатообіцяючі шляхи для цільових втручань. З усім тим, необхідні подальші поглиблені дослідження, щоб всебічно вивчити тубулоінтерстиціальні запальні ураження при DKD і зібрати надійні докази, що підтверджують ці цілі та шляхи. Приділяючи пріоритет лікуванню тубулоінтерстиціального запалення, ми можемо покращити наше розуміння ДЗН, розробити інноваційні стратегії профілактики та лікування та, зрештою, покращити результати та загальну якість життя людей, уражених цією хворобою.
ДЖЕРЕЛО: https://www.ncbi.nlm.nih.gov/

Щоб дати відповіді на запитання до цього матеріалу та отримати бали,
будь ласка, зареєструйтеся або увійдіть як користувач.
Реєстрація
Вхід
Матеріали з розділу

Вплив використання циклоспорину та метотре ...
.jpg)
Блокаторы ренин-ангиотензиновой системы, с ...

Запис. День 2. «UFMF 2025: сучасні проблем ...

Клінічне завдання. Підвищена втомлюваність ...

Фактори ризику гострої ниркової недостатно ...
Історичні нариси про вивчення сечовивідної ...
