Європейський день обізнаності про муковісцидоз
Дата публікації: 21.11.2023
Автори: Відкриті джерела , Редакція платформи «Аксемедін»
Ключові слова: скринінг, клінічна настанова, муковісцидоз, діагностика муковісцидозу

Щороку 21 листопада проводиться Європейський день обізнаності про муковісцидоз.
Муковісцидоз (МВ) — мультисистемне захворювання, спричинене патогенними мутаціями гена CFTR (трансмембранний регулятор провідності МВ). Типові симптоми та ознаки включають стійку легеневу інфекцію, недостатність підшлункової залози та підвищений рівень хлоридів поту. Однак у багатьох пацієнтів спостерігаються легкі або нетипові симптоми, тому клініцисти повинні бути уважними до можливості МВ, навіть якщо присутні лише деякі звичайні ознаки. Діагностика МВ базується на виявленні генетичних та/або функціональних аномалій гена CFTR.
.jpg)
Пропонуємо вам переглянути клінічну настанову щодо критеріїв постановки діагнозу муковісцидоз.
ЕПІДЕМІОЛОГІЯ
У Сполучених Штатах CF зустрічається приблизно у 1:3200 білих американців, 1:10 000 іспаномовних американців, 1:10 500 корінних американців, 1:15 000 чорношкірих американців і 1:30 000 азіатських американців. Муковісцидоз стає все більш визнаним у всьому світі, не тільки в Північній Америці, Європі та Австралії (регіони, найбільш знайомі з МВ), але також у Південній та Східній Азії, Африці та Латинській Америці, хоча відома поширеність у цих регіонах нижча. Оцінки поширеності, ймовірно, зростуть зі збільшенням визнання захворювання в усіх групах населення, використанням скринінгу новонароджених і збільшенням визнання осіб з легким захворюванням або захворюванням, обмеженим однією системою органів.
ВИЗНАЧЕННЯ
Муковісцидоз — класична або типова форма МВ діагностується, якщо пацієнт демонструє клінічне захворювання однієї або кількох систем органів (як описано нижче) і має підвищений рівень хлоридів поту (≥60 ммоль/л). Більшість із цих пацієнтів мають прояви захворювання в багатьох системах органів (верхніх і нижніх дихальних шляхів, підшлункової залози, шлунково-кишкового тракту та чоловічої репродуктивної системи).
Приблизно 2 відсотки пацієнтів відповідають діагностичним критеріям МВ, але їм не вистачає однієї чи кількох класичних ознак, описаних вище. Вони можуть мати легкі клінічні симптоми та/або нормальний або проміжний результат хлориду поту. Таким пацієнтам все ще може бути поставлений діагноз МВ, якщо вони відповідають генетичним або функціональним критеріям діагнозу, включаючи дві копії хвороботворної мутації в гені регулятора трансмембранної провідності CF (CFTR) на кожному батьківському алелі (тобто в транс) або аномальна назальна різниця потенціалів (NPD). Ці пацієнти з більшою ймовірністю з’являться пізніше в дитинстві чи дорослому віці та мають незвичайні мутації CFTR, які можуть не бути включеними до стандартної скринінгової панелі CF. У минулому ці фенотипи називали «некласичним» або «атиповим» CF, але зараз ці терміни не рекомендуються, оскільки вони є неточними та стосуються різних клінічних фенотипів.
Розлад, пов’язаний з CFTR. Розлад, пов’язаний з CFTR, визначається як клінічне захворювання, обмежене лише однією системою органів, пов’язане з певними ознаками дисфункції CFTR, що не відповідає повним генетичним або функціональним критеріям для діагностики МВ. Клінічні прояви можуть включати ізольовану обструктивну азооспермію, хронічний синусит, хронічний панкреатит або захворювання легенів, що проявляється у дорослому віці. У таких пацієнтів тестування може виявити лише одну хвороботворну мутацію в показниках CFTR і проміжних показниках хлориду поту та NPD. Ці пацієнти повинні пройти повне секвенування генів, включаючи оцінку генних дуплікацій або делецій, щоб підтвердити відсутність двох хвороботворних мутацій. Якщо присутні дві хвороботворні мутації, у пацієнта має бути діагностовано МВ, а не розлад, пов’язаний із СВТР.
За особами цієї категорії необхідно періодично спостерігати, щоб переконатися, що не виникають нові прояви захворювання, і їм необхідно отримати генетичну консультацію. Очікувана поширеність і прояви захворювання в осіб із розладом, пов’язаним з CFTR, можуть змінитися в майбутньому, оскільки методи діагностики мутацій і дисфункції CFTR стають більш чутливими та застосовуються ширше.
Відомо, що на інші захворювання впливає генотип CFTR, але вони не відповідають критеріям для діагностики МВ або розладу, пов’язаного з CFTR; вони залежать від не- CFTR генів і впливу навколишнього середовища на додаток до дисфункції CFTR. Наприклад, частота хронічного риносинуситу, бронхіту, бронхоектазів та алергічного бронхолегеневого аспергільозу підвищується серед осіб, у яких ідентифікована лише одна мутація CFTR.
CRMS/CFSPID — CFTR-пов’язаний метаболічний синдром (CRMS) — це термін, який описує немовлят і дітей із сумнівним діагнозом після скринінгу новонароджених на МВ і виявляється у 3–4 відсотків немовлят із позитивним результатом скринінгу новонароджених. Термін «МВ скринінг позитивний, непереконливий діагноз» (CFSPID) є еквівалентним і використовується в Європі.
CRMS/CFSPID описує безсимптомне немовля з позитивними результатами скринінгу новонароджених і:
● Проміжні результати хлориду поту (від 30 до 59 ммоль/л) у двох окремих випадках і менше двох мутацій, що викликають МВ
АБО
● Нормальні результати хлориду поту (≤29 ммоль/л) у двох окремих випадках і двох мутаціях CFTR, принаймні одна з яких не є чітко віднесеною до категорії, що викликає МВ

Оскільки природний перебіг немовлят із такими характеристиками непередбачуваний, CRMS/CFSPID є тимчасовим діагнозом і вимагає подальшого моніторингу та тестування в центрі, що має досвід МВ, включаючи принаймні один додатковий тест на хлорид поту. Деякі можуть продовжувати розвивати позитивний пітовий тест і клінічні характеристики МВ (хоча хвороба часто є легкою), інші можуть розвинути симптоми захворювання, пов’язаного з CFTR, включаючи ізольоване чоловіче безпліддя, а треті можуть залишатися повністю безсимптомними. Консенсусні заяви щодо оцінки та ведення немовлят із CRMS були опубліковані в Європі та Сполучених Штатах.
ОГЛЯД КЛІНІЧНИХ ОСОБЛИВОСТЕЙ
МВ спричинений мутаціями в білку регулятора трансмембранної провідності CF (CFTR), складному хлоридному каналі та регуляторному білку, який міститься у всіх екзокринних тканинах. Порушений транспорт хлоридів та/або інших іонів, на які впливає CFTR, таких як натрій і бікарбонат, призводить до густих, в’язких виділень у легенях, підшлунковій залозі, печінці, кишечнику та репродуктивному тракті та до збільшення вмісту солі в секреті потових залоз. У типового пацієнта з муковісцидозом розвивається мультисистемне захворювання, що залучає кілька або всі ці органи.

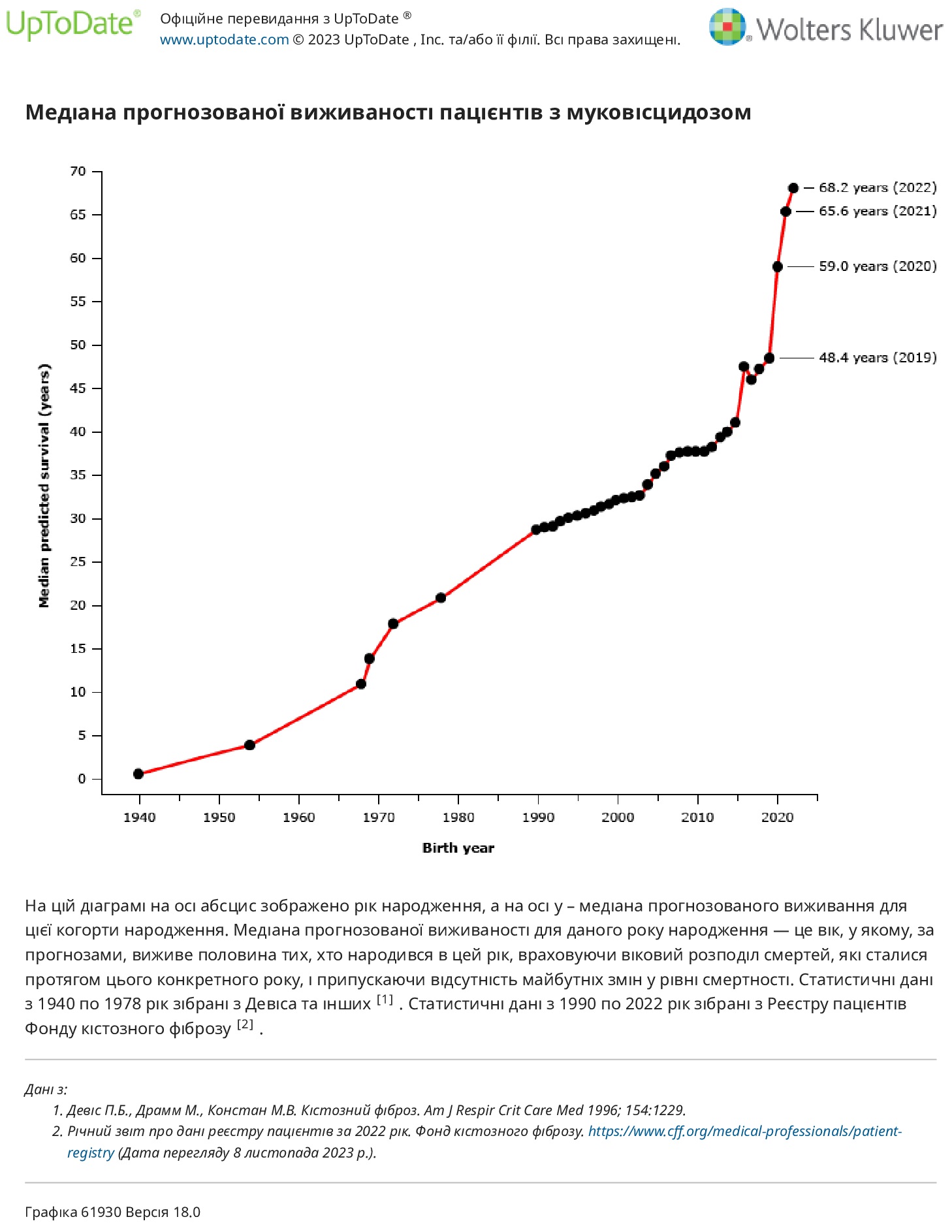
Симптоми — у минулому більшості пацієнтів діагностували МВ після появи симптомів. Через розширення програм скринінгу новонароджених протягом останніх 20 років відбулося різке збільшення кількості випадків МВ, виявлених до появи симптомів. У 2001 році менше 10 відсотків випадків МВ у Сполучених Штатах були діагностовані на основі програм скринінгу новонароджених. До 2021 року 64,4 відсотка від загальної кількості нових діагнозів МВ і 93,8 відсотка діагнозів серед немовлят віком до шести місяців були виявлені за допомогою скринінгу новонароджених. Існують докази того, що особи, яким діагностовано до появи симптомів, мають кращу функцію легенів, показники нейрокогнітивного тестування та харчові результати пізніше в житті та менше використовують ресурси охорони здоров’я.
Пренатальні знахідки — Деякі випадки муковісцидозу супроводжуються аномальними результатами рутинного пренатального ультразвукового дослідження, включаючи гіперехогенний кишечник. Ризик МВ найвищий, якщо є ознаки меконіального перитоніту (розсіяні кальцифікати спостерігаються по очеревині плода), розширення кишечника або відсутність жовчного міхура. Якщо ці результати є на УЗД плода, ми пропонуємо запропонувати батькам пренатальний скринінг на носійство МВ.
На додаток до специфічних даних, описаних вище, МВ може бути пов’язаний з передчасними пологами та низькою вагою при народженні. У великому реєстровому дослідженні вплив МВ на вагу при народженні оцінювався приблизно як -200 г; лише 40 відсотків цього впливу на вагу при народженні було пояснено більш раннім терміном вагітності. Механізми цих впливів на ріст плоду не встановлені.
Симптоматичні прояви у немовлят і дітей. До впровадження широкого скринінгу новонароджених у Сполучених Штатах і багатьох інших країнах у немовлят і дітей зазвичай діагностували муковісцидоз після появи одного або кількох з наступних симптомів:
● Меконіальний ілеус – 20% пацієнтів
● Респіраторні симптоми – 45% пацієнтів
● Відсутність розвитку – 28% пацієнтів
Для немовлят з меконієвою кишковою непрохідністю середній вік встановлення діагнозу становив два тижні. Для тих, хто має інші симптоми, середній вік встановлення діагнозу становив 14,5 місяців (міжквартильний діапазон від 4,2 до 65 місяців). Ці клінічні прояви все ще актуальні для популяцій, які не проходять рутинний скринінг новонароджених на МВ.
Немовлята з важкою нелікованою недостатністю підшлункової залози іноді мають синдром набряку з гіпопротеїнемією, втратою електролітів, анемією, алопецією, дерматитом і затримкою розвитку через порушення всмоктування та недоїдання, включаючи дефіцит незамінних жирних кислот, цинку, вітамінів і білків.
Симптоматичний прояв у зрілому віці — Пацієнти, які мають муковісцидоз у більш пізньому віці, частіше мають атипові симптоми. Одне велике ретроспективне когортне дослідження понад 1000 пацієнтів з муковісцидозом показало, що у 7 відсотків діагноз був поставлений у віці ≥18 років. Пацієнти з діагнозом у дорослому віці частіше, ніж діти, мали шлунково-кишкові симптоми, цукровий діабет і безпліддя. Крім того, дорослі з муковісцидозом частіше, ніж діти, мали незвичайні генетичні мутації, нормальну функцію підшлункової залози та сумнівні результати тестів на хлорид поту.
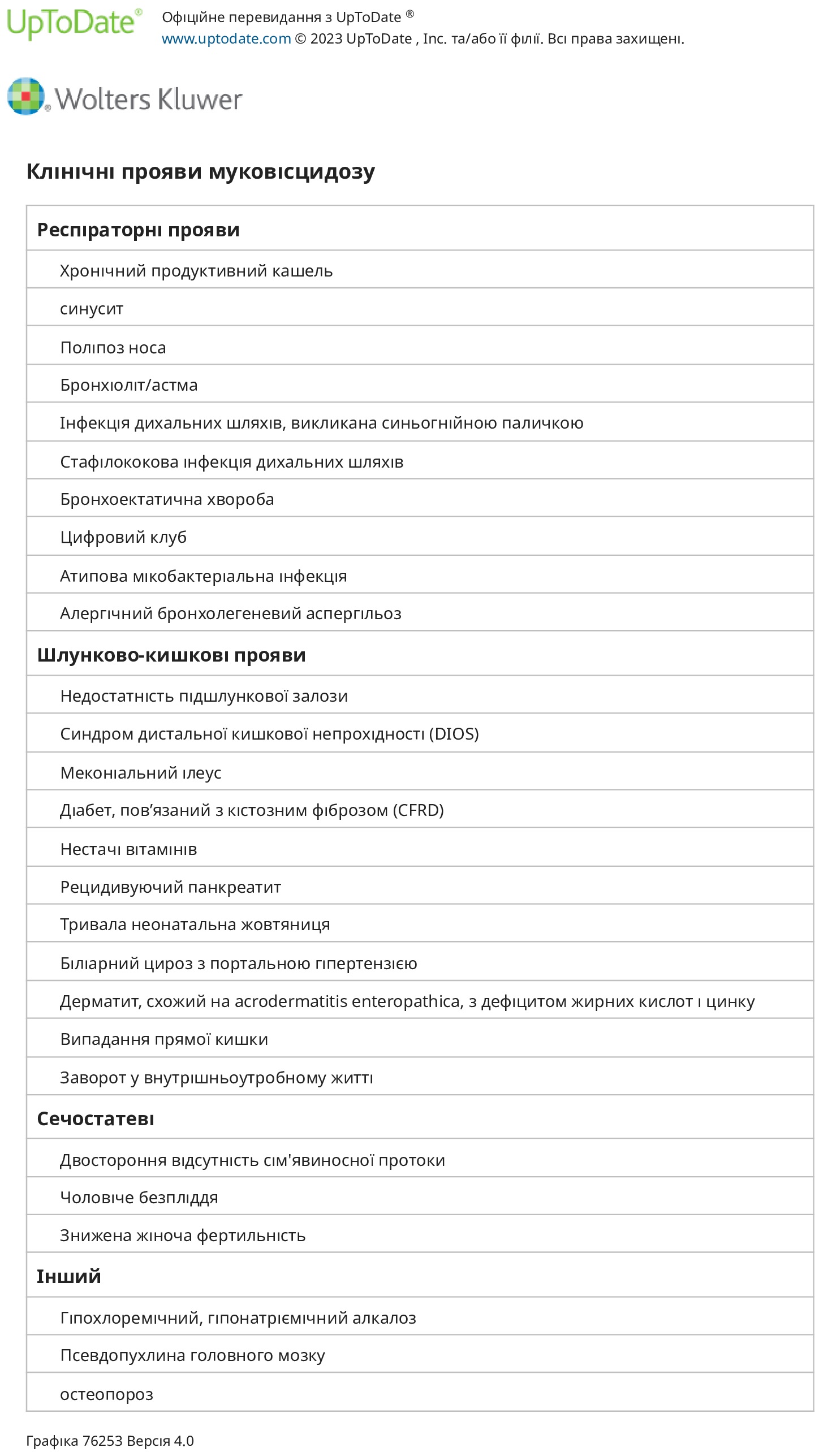
Ураження дихальних шляхів — Типові респіраторні прояви муковісцидозу включають постійний продуктивний кашель, гіперінфляцію легеневих полів на рентгенограмі грудної клітки та тести легеневої функції, які відповідають обструктивному захворюванню дихальних шляхів. Початок клінічних симптомів значно варіюється через відмінності в генотипі CFTR та інших індивідуальних факторів, але порушення легеневої функції часто можна виявити навіть за відсутності симптомів. Прогресування захворювання включає гострі загострення з кашлем, тахіпное, задишкою, збільшенням виділення мокротиння, нездужанням, анорексією та втратою ваги. Ці явища пов’язані з гострою тимчасовою втратою функції легень, яка покращується під час лікування, але з часом призводить до постійної втрати функції легень.
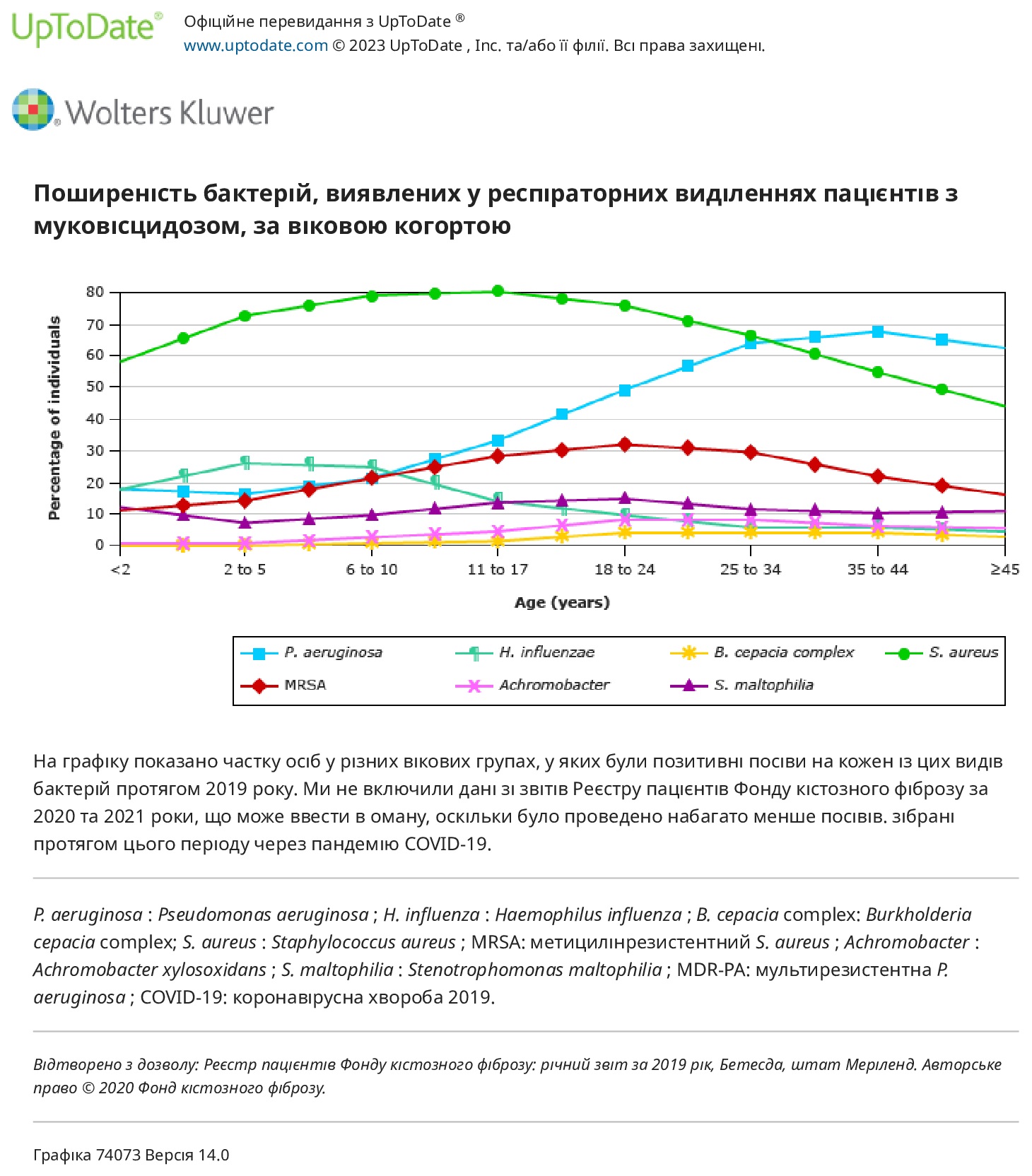
Транзиторна інфекція дихальних шляхів патогенними бактеріями часто виникає на ранньому етапі життя. Врешті-решт, протягом багатьох років і коливається в різних індивідів, встановлюється хронічна інфекція дихальних шляхів або Staphylococcus aureus, або грамнегативними бактеріями, часто з рентгенографічними ознаками бронхоектазів. S. aureus і нетипова Haemophilus influenzae є поширеними збудниками в ранньому дитинстві, але Pseudomonas aeruginosa остаточно виділяють із респіраторних виділень більшості пацієнтів. S. aureus, особливо повільно зростаючий або варіанти «маленької колонії», продовжує спричиняти значну захворюваність у дітей старшого віку та дорослих з МВ. Інші мікроби, до колонізації та інфікування яких схильні пацієнти з МВ, включають Stenotrophomonas maltophilia, Achromobacter xylosoxidans, Burkholderia cepacia complex, нетуберкульозні мікобактерії (особливо Mycobacterium avium complex і Mycobacterium abscessus) і нитчастий гриб Aspergillus fumigatus. Ця схильність до П. інфекція aeruginosa може бути частково спричинена порушенням кліренсу, безпосередньо спричиненим дефектом CFTR.
Успіхи в лікуванні, включаючи використання модуляторів CFTR, призвели до різкого збільшення прогнозованої виживаності для осіб з МВ.
Захворювання пазух і носоглотки — у більшості пацієнтів з МВ розвивається захворювання пазух. Рентгенографія виявляє панопацифікацію навколоносових пазух у 90-100 відсотків пацієнтів старше восьми місяців. Поліпоз носа спостерігається у 10-32 відсотків пацієнтів і спричинений хронічним риносинуситом. Клінічні прояви поліпів носа/носових пазух варіюються від безсимптомних виявлень під час фізикального огляду до легких або важких симптомів, включаючи ринорею, закладеність носа, закладеність носа, постназальну крапельницю, хропіння, обструктивне апное сну, гіпоназальну мову, носову кровотечу, гіпосмію/аносмію, агевзію, біль у обличчі. біль або головний біль, біль над верхніми зубами, відчуття тиску на чолі та обличчі, двоїння в очах і розширення носової піраміди. Захворювання носових пазух може проявлятися хронічною закладеністю носа, головними болями, кашлем, викликаним хронічним постназальним виділенням, і порушенням сну. Інфекції пазух можуть спровокувати загострення нижніх дихальних шляхів у деяких пацієнтів, хоча мікроорганізми, виявлені в пазухах, не завжди збігаються з тими, що виділяються з легенів. Перебіг захворювання синусів змінився в епоху лікування модуляторами CFTR. До появи модуляторів CFTR лікування захворювань носових пазух і поліпозу носа зазвичай було незадовільним, пацієнти рідко отримували тривале полегшення симптомів. З початком терапії модулятором CFTR багато людей з CF відчувають стійке полегшення хронічного риносинуситу та поліпозу носа.
Захворювання підшлункової залози — МВ асоціюється з кількома різними типами захворювань підшлункової залози:
● Недостатність підшлункової залози. Недостатність зовнішньої секреції підшлункової залози присутня від народження приблизно у двох третин пацієнтів із МВ. Загальні симптоми та ознаки недостатності підшлункової залози включають стеаторею, яка характеризується частим, об’ємним стільцем з неприємним запахом, який може бути маслянистим, а також затримкою розвитку або низьким набором ваги через порушення всмоктування жиру та білка. У пацієнта з чітким діагнозом МВ діагноз недостатності підшлункової залози зазвичай може бути встановлений на основі цих клінічних симптомів, клінічної відповіді на замісну терапію ферментами підшлункової залози та/або лабораторних досліджень (наприклад, фекальна еластаза). У немовлят із важкою нелікованою недостатністю підшлункової залози іноді спостерігаються синдром набряку, гіпопротеїнемія, втрата електролітів та анемія внаслідок порушення всмоктування макро- та мікроелементів. У таких пацієнтів також можуть виникати симптоми, спричинені дефіцитом жиророзчинних вітамінів A, D, E та K. Дефіцит вітаміну K може проявлятися коагулопатією, а дефіцит вітаміну D — рахітом.
● Панкреатит – порушення секреції проток і ацинарів підшлункової залози викликає прогресуюче ураження підшлункової залози. Це може призвести до гострого або рецидивуючого панкреатиту. Панкреатит розвивається приблизно у 10 відсотків пацієнтів із муковісцидозом із недостатністю підшлункової залози, але рідко серед пацієнтів із симптоматичною недостатністю підшлункової залози.
● Діабет, пов’язаний з МВ (CFRD) – у пацієнтів із екзокринною недостатністю підшлункової залози часто розвивається дисфункція ендокринної підшлункової залози, що призводить до непереносимості глюкози та CFRD. Приблизно у 25 відсотків пацієнтів розвивається CFRD до 20 років, і до 50 відсотків дорослих з МВ мають CFRD.
Меконієвий ілеус і дистальна непрохідність клубової кишки. Меконіальний ілеус характеризується закупоркою кишечника меконієм у новонародженого. Це проблема, яка виникає у 10-20 відсотків новонароджених з МВ. Навпаки, від 80 до 90 відсотків немовлят з меконієвою кишковою непрохідністю мають МВ, хоча недоношені немовлята можуть частіше мати меконіальну кишкову непрохідність без МВ. Меконіальна кишкова непрохідність може виникнути у пацієнтів із різноманітними мутаціями CFTR. Висока частота сімейних рецидивів свідчить про те, що інші генетичні модифікатори сприяють розвитку меконіальної кишкової непрохідності. Приблизно в 40% випадків супроводжується перфорацією або атрезією тонкої чи клубової кишки.
Епізоди непрохідності тонкої кишки також можуть виникати у дітей і дорослих. Вони відомі як дистальний інтестинальний обструктивний синдром (DIOS), і їх слід враховувати у будь-якого пацієнта з МВ, який має абдомінальний біль. Випадання прямої кишки — випадання прямої кишки зараз рідко трапляється у дітей з МВ. Схоже, що це пов’язано із запорами та/або недоїданням і більш імовірно, якщо ефективна терапія ферментами підшлункової залози не встановлена. У минулому пролапс прямої кишки був набагато більш поширеним явищем у 20% пацієнтів із класичним муковісцидозом, мабуть, через пізнішу діагностику та, можливо, неоптимальне лікування ферментною терапією підшлункової залози порівняно з сучаснішою практикою.
Гепатобіліарне захворювання — вогнищевий біліарний цироз печінки, спричинений застійною жовчю, присутній у багатьох пацієнтів і може спричинити підвищення рівня лужної фосфатази в сироватці крові та часточкову гепатомегалію. Безсимптомне захворювання печінки є загальним виявленням при розтині. У меншості пацієнтів захворювання печінки прогресує з перипортальним фіброзом, цирозом, симптоматичною портальною гіпертензією, секвестрацією селезінки та варикозною кровотечею. МВ є третьою основною причиною трансплантації печінки в пізньому дитинстві.
Безпліддя. Понад 95 відсотків чоловіків із МВ є безплідними через дефекти транспорту сперми, хоча сперматогенез не впливає.. Більшість із цих чоловіків мають неповністю розвинені вольфові структури, найчастіше, відсутність сім’явивідної протоки. Ці аномалії, ймовірно, відображають критичну роль CFTR в органогенезі цих структур. Майже половина всіх чоловіків із вродженою двосторонньою відсутністю сім’явиносної протоки та нормальною функцією легень мають дві мутації CFTR.
Мікрохірургічна аспірація сперматозоїдів епідидиму та інтрацитоплазматична ін’єкція сперматозоїдів можуть дозволити постраждалим чоловікам стати біологічними батьками.
Жінки з МВ виявляються менш фертильними, ніж нормальні здорові жінки. Зменшення фертильності викликане в основному недоїданням і виробленням аномально міцного цервікального слизу. З усім тим, завжди слід припускати, що жінки з муковісцидозом можуть завагітніти, і пацієнтам слід відповідним чином консультувати щодо контрацепції та рішень про народження дитини. Коли пацієнтки з муковісцидозом завагітніли, наслідки для матері та плода загалом сприятливі, якщо об’єм форсованого видиху за одну секунду (ОФВ1) до вагітності перевищує 50–60 відсотків прогнозованого значення.
Лікування за допомогою модуляторної терапії CFTR, очевидно, підвищує показники фертильності. Ретельне генетичне консультування має важливе значення для потенційних батьків з МВ, оскільки всі нащадки таких осіб будуть носіями мутацій МВ, і ризик дітей, уражених МВ, є високим.
Розлади опорно-рухового апарату — у пацієнтів із муковісцидозом знижений мінеральний вміст кісткової тканини (остеопенія та остеопороз) і підвищена частота переломів і кіфосколіозу. Клінічно значуще зниження щільності кісткової тканини спостерігається до 30 відсотків пацієнтів із МВ у всіх вікових групах і до 75 відсотків дорослих із МВ. Кілька різних механізмів, очевидно, сприяють захворюванню кісток, включаючи порушення всмоктування вітаміну D, поганий статус харчування, відсутність фізичної активності, терапію глюкокортикоїдами та затримку статевого дозрівання або гіпогонадизм. Обмежені дані свідчать про те, що пацієнти, гомозиготні за мутацією F508del, мають особливо високий ризик зниження мінеральної щільності кісткової тканини. Окремо розглядаються патогенез, оцінка та профілактика захворювань кісток у хворих на МВ.
Гіпертрофічна остеоартропатія — це синдром, що характеризується аномальною проліферацією шкіри та кісткової тканини в дистальних частинах кінцівок, що виникає у зв’язку з рентгенологічно підтвердженим утворенням нової кістки в окістя. Булини пальців і гіпертрофічна остеоартропатія є різними проявами одного і того ж процесу захворювання. Хоча булавоподібні пальці на руках і ногах часто зустрічаються у пацієнтів із тривалим МВ, гіпертрофічна остеоартропатія зустрічається рідко (5 відсотків пацієнтів).
Артропатія, пов’язана з CF, зустрічається у 2-9 відсотків пацієнтів і характеризується короткими епізодами болю та набряку суглобів. Ці ознаки іноді супроводжуються хворобливими вузловими ураженнями шкіри та пурпурою.
Рецидивуючий венозний тромбоз — МВ є фактором ризику повторного венозного тромбозу, ймовірно, частково через часту потребу в центральному венозному катетері (CVC).
Анемія. Пацієнти з муковісцидозом знаходяться в групі ризику анемії, яка присутня приблизно у 10 відсотків дітей і частіше зустрічається з віком і зниженням легеневої функції. Іноді анемія є первинною ознакою у немовляти. Механізми анемії включають:
● Дефіцит заліза, викликаний порушенням метаболізму заліза. У дітей це, здається, не пов’язано з поганим станом харчування, і механізм, за допомогою якого виникає дефіцит заліза, залишається неясним.
● Анемія хронічного запалення (також відома як анемія хронічного захворювання) внаслідок хронічного та гострого захворювання легень.
● Дефіцит заліза через крововтрату, як у пацієнтів з кровохарканням або варикозним розширенням вен стравоходу чи шлунка.
● Іншими причинами в деяких випадках є ниркова недостатність або пригнічення кісткового мозку у пацієнтів після трансплантації.
Визначення причини або причин анемії в окремого пацієнта може бути складним завданням. У пацієнтів із запаленням легень сироватковий феритин може бути помилково нормальним або підвищеним. Хронічна гіпоксемія є фізіологічним тригером для синтезу гемоглобіну. Таким чином, пацієнти з хронічною гіпоксемією та нормальним гемоглобіном можуть вважатися такими, що мають «відносну» анемію, що відображає основний дефіцит заліза або інше порушення синтезу гемоглобіну.
Порушення електролітного балансу — іноді в осіб з МВ може розвинутися підгостра або хронічна гіповолемія з гіпонатріємією, гіпохлоремією, гіпокаліємією та метаболічним алкалозом (іноді відомий як синдром псевдо-Барттера).
Цей стан спричинений надмірною втратою натрію та хлориду з потом і може розвинутися у пацієнтів із МВ із недостатнім споживанням натрію. Немовлята особливо схильні до ризику, оскільки вміст солі в грудному молоці або дитячій суміші може бути недостатнім, і необхідна добавка натрію. Іноді це основна ознака МВ. Захворювання також може розвинутися у дітей старшого віку або дорослих під час теплового стресу.
Нефролітіаз і нефрокальциноз. Нефролітіаз і нефрокальциноз є поширеними у пацієнтів з МВ.. Ентеральна гіпероксалурія (через порушення всмоктування жиру внаслідок зниження секреції панкреатичних ферментів) і гіпоцитратурія (через хронічний метаболічний ацидоз) є ймовірними факторами ризику.
Аквагенні зморшки — Аквагенні зморшки на долонях (зморшки та вузлики, які розвиваються після кількох хвилин занурення у воду пов’язані з мутаціями CFTR.
СКРИНІНГ НОВОНАРОДЖЕНИХ
Обґрунтування скринінгу новонароджених полягає в тому, що раннє виявлення МВ може призвести до більш раннього втручання та покращення результатів, оскільки уражених осіб діагностують, направляють і лікують раніше в житті порівняно з особами, у яких діагностовано МВ після прояву симптомів МВ.
На додаток до можливості раннього виявлення та лікування осіб з МВ, неонатальний скринінг ідентифікує популяцію для вивчення механізму раннього (доклінічного) ураження легень. У майбутньому можна буде оцінити ефективність нових методів лікування для відстрочення або запобігання розвитку легеневих захворювань у цих пацієнтів із доклінічним захворюванням.
Методи — Скринінг новонароджених зазвичай використовує два серійних аналізи; немовлят з ненормальними результатами першого аналізу перевіряють повторно за допомогою другого аналізу. Два аналізи, які використовуються для скринінгу новонароджених, це сироватковий імунореактивний трипсиноген (IRT) і аналіз дезоксирибонуклеїнової кислоти (ДНК) на мутації в гені регулятора трансмембранної провідності CF (CFTR). IRT використовується як початковий скринінговий тест у багатьох протоколах і супроводжується другим тестом IRT на іншому зразку немовляти (протокол IRT/IRT) або тест ДНК на початковому зразку (протокол IRT/DNA). Протокол скринінгу IRT/IRT має дещо нижчу вартість, але більше затримок або пропусків діагностики порівняно з протоколом скринінгу IRT/ДНК.
Немовлята з позитивними результатами скринінгу новонароджених на МВ повинні пройти тестування на хлорид поту, щоб визначити, чи є у них МВ. Для оптимальної точності тестування поту слід проводити, коли немовля виповнилося принаймні два тижні та важить понад 2 кг, використовуючи лабораторні методи, передбачені Фондом кістозного фіброзу.
ДІАГНОСТИКА
Діагноз МВ ґрунтується на сумісних клінічних результатах з біохімічним або генетичним підтвердженням. Тест на хлорид поту є основою лабораторного підтвердження, хоча тести на специфічні мутації, назальну різницю потенціалів (NPD), імунореактивний трипсиноген (IRT), фекальний жир у калі або секрецію ферментів підшлункової залози також можуть бути корисними в деяких випадках.

Діагностичні критерії — Обидва наступні критерії повинні відповідати для діагностики МВ:
● Клінічні симптоми, що відповідають МВ принаймні в одній системі органів, або позитивний скринінг новонароджених або наявність брата або сестри з CFІ
● Докази дисфункції трансмембранного регулятора провідності (CFTR) (будь-який із наведених нижче):
- Підвищений рівень хлориду поту ≥60 ммоль/л
- Наявність двох хвороботворних мутацій у гені CFTR, по одній від кожного батьківського алеля
- Аномальний NPD
Точність вимірювань хлориду поту та NPD залежить від оператора, тому дуже важливо, щоб тестування проводилося в досвідчених центрах, дотримуючись стандартних вказівок.
Лабораторне тестування. Докази дисфункції CFTR можна отримати за допомогою аналізу хлориду поту, молекулярного тестування на мутації гена CFTR або вимірювання NPD. У більшості випадків визначення хлориду поту є першим і найважливішим тестом. Тест ДНК використовується для підтвердження або для подальшого дослідження пацієнтів із проміжними результатами хлориду поту, а також для прогностичних та епідеміологічних цілей у осіб з позитивними результатами тесту на хлорид поту. Діагностичний підхід викладено в алгоритмі.
Хлорид поту — Тест на хлорид поту залишається основним тестом для діагностики МВ; тестування поту виконується шляхом збору поту за допомогою іонофорезу пілокарпіну та хімічного визначення концентрації хлориду.
Показання — для уточнення діагнозу МВ пацієнти з такими характеристиками повинні пройти аналіз поту:
● Немовлята з позитивними результатами скринінгу новонароджених на муковісцидоз (виконують після двотижневого віку та >2 кг, якщо немає симптомів)
● Немовлята з симптомами, що вказують на МВ (наприклад, меконіальний ілеус)
● Діти старшого віку та дорослі з симптомами, що вказують на МВ (наприклад, чоловіче безпліддя, хронічні респіраторні інфекції або хронічний синусит)
● Брати й сестри пацієнта з підтвердженим МВ, якщо діагноз неможливо встановити на основі генетичного тестування
Інтерпретація — Інтерпретація результатів хлориду поту така:
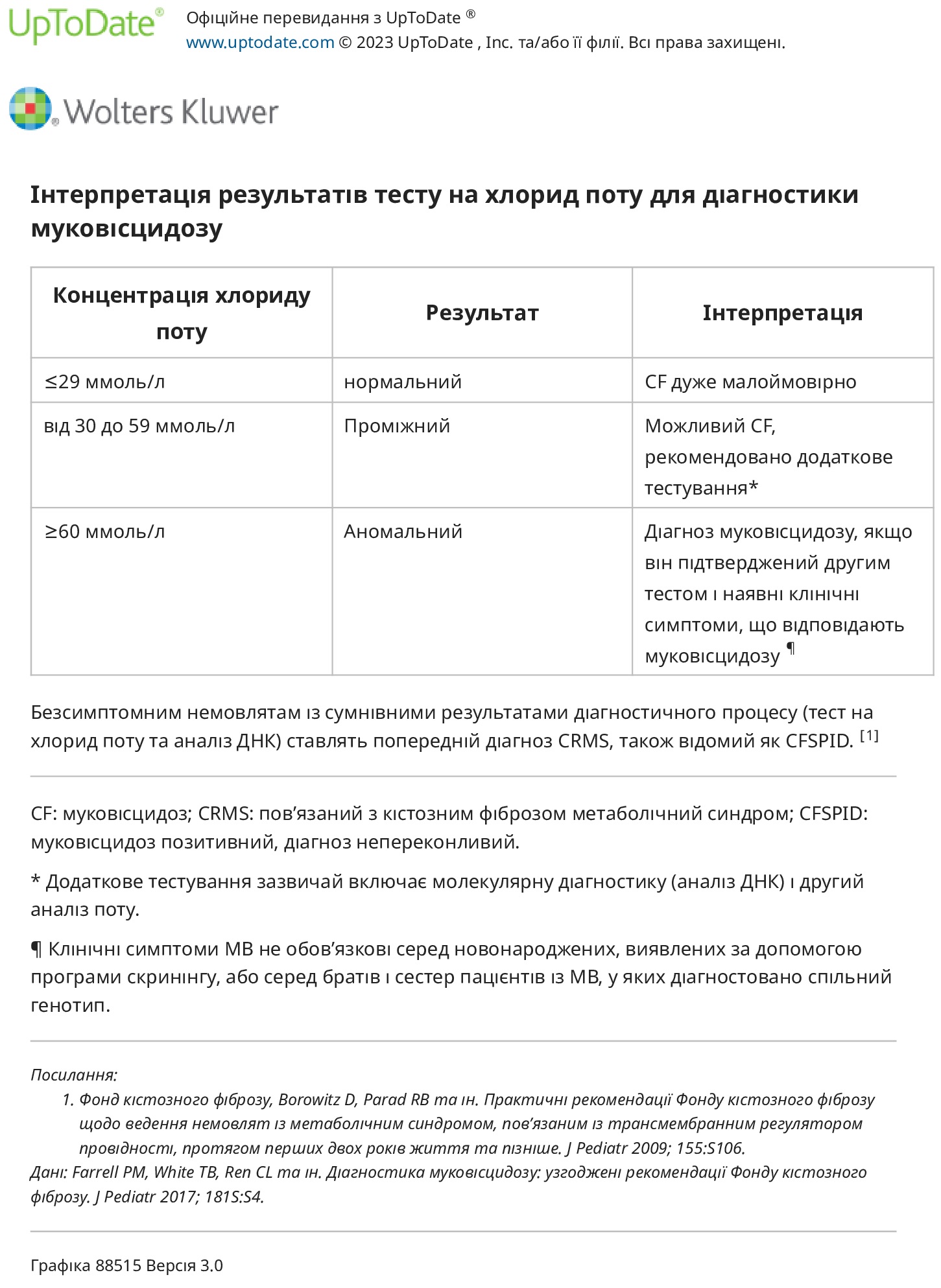
● Норма – Хлорид поту ≤29 ммоль/л є нормальним. Цього результату достатньо, щоб виключити МВ у більшості осіб.
● Проміжний – Хлорид поту від 30 до 59 ммоль/л є проміжним. Цей результат свідчить про можливу МВ і вимагає подальшої оцінки шляхом повторного тестування хлориду поту, а також секвенування CFTR. Приблизно 20 відсотків дітей із проміжними результатами хлориду поту матимуть докази ДНК МВ при розширеному аналізі.
Для безсимптомних немовлят із проміжними результатами тест на хлорид поту слід повторити у віці від одного до двох місяців, а потім з інтервалом у 6-12 місяців, доки діагноз не буде чітким. У немовлят чи дітей із симптомами або у немовлят віком до двох тижнів на момент першого тестування може бути доречним повторити тест на хлориди поту раніше.
● Ненормальний – рівень хлориду поту ≥60 ммоль/л є ненормальним. У разі повторного підтвердження цього достатньо для підтвердження діагнозу МВ у пацієнтів із клінічними симптомами МВ. Клінічні симптоми не потрібні для немовлят, які були виявлені під час скринінгу новонароджених.
Необхідно також враховувати ймовірність станів, не пов’язаних із МВ, які можуть бути пов’язані з підвищеним вмістом хлоридів поту. Визначення генотипу CFTR є важливим, оскільки результати можуть вплинути на вибір лікування, а також підтвердити діагноз. Нові стратегії лікування спрямовані на конкретні мутації CFTR, використовуючи модулятори CFTR.

Безсимптомним немовлятам із сумнівними результатами діагностичного процесу (тест на вміст хлориду поту та аналіз ДНК) ставлять попередній діагноз метаболічний синдром, пов’язаний із CFTR (CRMS)/позитивний, непереконливий діагноз (CFSPID). Це пояснюється тим, що результати та симптоми хлориду поту можуть змінюватися з віком, а клінічні наслідки деяких варіантів гена CFTR неясні. Цих немовлят слід направити на повніше обстеження та подальше спостереження, включаючи принаймні один повторний тест на вміст хлориду поту в акредитованому центрі МВ, клінічну оцінку досвідченим клініцистом МВ і, можливо, розширене генетичне тестування або функціональний аналіз (наприклад, вимірювання NPD).
Техніка — Дослідження поту виконується шляхом збору поту за допомогою іонофорезу пілокарпіну та хімічного визначення концентрації хлориду. Тест виконується шляхом нанесення пілокарпіну на шкіру, щоб стимулювати утворення поту. Невелика система збору наноситься на те саме місце на поверхню шкіри, і все покривається поліетиленовою плівкою, щоб сприяти потовиділенню та забезпечити збір поту. Зразки повинні бути зібрані та перевірені в двох примірниках, якщо це можливо, для забезпечення якості. Достатня кількість рідини зазвичай виділяється протягом приблизно години. Тест безболісний, але складний для правильного виконання.
Молекулярна діагностика. Молекулярна діагностика є стандартною частиною скринінгу новонароджених у всіх Сполучених Штатах, хоча конкретна форма та час аналізу різняться. Якщо виявлено дві мутації, що викликають МВ, і аналіз поту є проміжним або позитивним, діагноз МВ підтверджується. Якщо дві мутації, що викликають CF, не виявлені, слід повторити аналіз поту. Якщо МВ неможливо остаточно діагностувати або виключити, немовляті ставлять попередній діагноз CRMS, і слід провести подальшу генетичну оцінку.
● Панелі генетичного скринінгу – Скринінг новонароджених зазвичай проводиться за допомогою панелей мутацій гена CFTR. Мутації, включені в панель, відрізняються в різних штатах залежно від етнічного різноманіття їхнього населення. Більшість штатів перевіряють щонайменше 23 найпоширеніші мутації, використовуючи панель, розроблену для популяційного скринінгу Американським коледжем медичної генетики (ACMG). Комісія ACMG ідентифікує приблизно 90 відсотків мутацій, що викликають МВ, у загальній популяції (і 97 відсотків мутацій у родинах ашкеназького єврейського походження).

● Секвенування генів – Секвенування CFTR слід проводити особам із будь-якою невизначеністю в діагнозі, зокрема:
- Пацієнти з проміжними результатами хлориду поту (на додаток до повторного визначення хлориду поту).
- Пацієнти з підтвердженою або підозрюваною МВ, якщо генотип ще невідомий. У цих пацієнтів секвенування генів підтверджує діагноз, а знання про специфічну мутацію CFTR також має важливі наслідки для лікування та прогнозу
- Пацієнти з нормальними результатами хлориду поту, якщо є сильна клінічна підозра на МВ.
ДИФЕРЕНЦІЙНА ДІАГНОСТИКА
Симптоми наступних захворювань можуть імітувати симптоми МВ:
● Первинні імунологічні аномалії, включаючи важкий комбінований імунодефіцит і загальний варіабельний імунодефіцит, можуть проявлятися при рецидивуючих синолегеневих інфекціях, подібних до МВ
● Первинна циліарна дискінезія також викликає рецидивуючі сино-легеневі інфекції, а також чоловіче безпліддя. Постраждалі пацієнти також схильні до рецидивів середнього отиту, і 50 відсотків мають situs inversus.
● Синдром Швахмана-Даймонда може викликати недостатність підшлункової залози, але зустрічається значно рідше, ніж МВ. Захворювання пов’язане з хронічними або рецидивуючими гематологічними аномаліями.
● Окремо розглядаються причини бронхоектазів у дітей, не пов’язані з МВ.
● Дефіцит альфа-1 антитрипсину може проявлятися різними фенотипами захворювань печінки та легенів, залежно від варіанту.
Діагностичну оцінку цих розладів слід розглянути, коли діагноз очевидної МВ не може бути підтверджений лабораторними дослідженнями.
ДЖЕРЕЛО: https://www.uptodate.com/

Щоб дати відповіді на запитання до цього матеріалу та отримати бали,
будь ласка, зареєструйтеся або увійдіть як користувач.
Реєстрація
Вхід
Матеріали з розділу
Персоніфікований підхід до лікування хроні ...

Клінічні випадки. Лінійний бульозний дерма ...

Клінічна настанова: Дефіцит вітамінів, пов ...

Клінічні випадки. Синдром чужої руки

Клінічна настанова: Лікування важкого рефр ...
Клінічний випадок. Біль у пальці
