: Метаболізм і транспорт ліків 100 найбільш прописаних пероральних препаратів
Дата публікації: 11.10.2023
Автори: Відкриті джерела , Редакція платформи «Аксемедін»
Ключові слова: ліки, препарати, метаболізм, взаємодія, фармакокінетика, фармакодинаміка
Безпечне та ефективне використання ліків вимагає розуміння метаболізму та транспорту. Ми визначили 100 препаратів, які найчастіше призначають у шести країнах, і провели пошук літератури за даними in vitro, щоб оцінити внесок ферментів фази I і II і транспортерів ліків у метаболізм і транспорт.
Вісімдесят дев’ять зі 100 ліків піддаються метаболізму або є відомими субстратами для переносників ліків. Ферменти фази I беруть участь у метаболізмі 67 ліків, тоді як ферменти фази II опосередковують метаболізм 18 ліків. CYP3A4/5 є найважливішим ферментом фази I, який бере участь у метаболізмі 43 препаратів, за ним йдуть CYP2D6 (23 препарати), CYP2C9 (23 препарати), CYP2C19 (22 препарати), CYP1A2 (14 препаратів) і CYP2C8 (11 препаратів). Більше половини ліків (54 препарати) є відомими субстратами для переносників ліків. Відомо, що Р-глікопротеїн (P-gp) бере участь у транспортуванні 30 ліків, тоді як білок резистентності раку молочної залози (BCRP) сприяє транспортуванню 11 ліків. Значна частина лікарських засобів піддається комбінації метаболізму фази I, метаболізму фази II та/або транспорту ліків.
Рекомендуємо долучитись до семінару «Поліморбідний пацієнт: раціональний підхід до лікування в амбулаторних умовах» від провідного спеціаліста Чернявського Володимира Володимировича.
Автори прийшли до висновку, що більшість препаратів, які найчастіше призначають, залежать від метаболізму або транспорту ліків.
Використання ліків зростає у всьому світі. Датське дослідження показало, що 51% осіб віком ≥75 років призначають п’ять або більше різних ліків. Ферментативний метаболізм і транспортування ліків важливі для всмоктування, розподілу, метаболізму та виведення ліків в організмі. Відомо, що метаболізм і транспортування ліків відрізняються як між людьми, так і всередині них, і це є проблемою, оскільки це спричиняє різну ефективність і токсичність лікування. Варіації в метаболізмі та транспортуванні ліків можуть бути спричинені взаємодією між ліками, епігенетикою, генетичними поліморфізмом в генах, які кодують ферменти, що метаболізують ліки, або транспортери ліків [наприклад, цитохром P450 (CYP) 2D6, CYP2C9 і CYP2C19 ], або внутрішні (наприклад, стать і запалення) і зовнішні фактори (наприклад, дієта та вплив хімічних речовин з навколишнього середовища).
Попереднє літературне дослідження описувало ферменти фази I, які беруть участь у метаболізмі 200 найбільш прописаних препаратів у Сполучених Штатах. У цьому документі надається оновлена карта метаболізму фази I, але також включаємо метаболізм фази II та транспортери ліків, що мають відношення до 100 препаратів, які найчастіше призначають у п’яти європейських країнах та Австралії. Автори включили дані з шести країн, щоб зробити список більш інтернаціональним. Крім того, узагальнили загальний внесок ферментів і транспортерів, щоб зрозуміти кількість ліків, на які вплинуть зміни в метаболізмі або транспорті ліків. Розглянули основні аспекти з літератури щодо найважливіших ферментів фази I та II та транспортерів ліків.
Визначення 100 найпопулярніших ліків, які призначають :
Автори отримали дані про вживання рецептурних ліків з шести країн: Данії, Швеції, Норвегії, Англії, Шотландії та Австралії. Дані були доступні за конкретними запитами до співробітників у шести країнах. Для кожної країни ми визначили препарати, які найчастіше призначають. Ранжування препаратів в окремих списках ґрунтувалося на різних показниках. Дані з Данії та Швеції базувалися на кількості рецептів на 1000 громадян, дані з Норвегії базувалися на кількості унікальних осіб, які купували ліки, дані з Англії та Шотландії базувалися на кількості відпущених товарів, а дані з Австралії були виходячи з кількості рецептів. Ліки, які містять дві діючі речовини, були розділені на окремі компоненти; наприклад, комбінація кодеїну та парацетамолу зараховувалась як використання обох препаратів окремо. Ми включили лише пероральні препарати для системного лікування, таким чином виключивши інгаляційні препарати, препарати для внутрішньовенного введення, дерматологічні препарати, препарати, які не всмоктуються з кишечника та дієтичні добавки. Були доступні лише дані про рецепти з аптек, таким чином виключаючи дані про рецепти з лікарень. Оскільки норвезькі дані містили найменшу кількість окремих ліків (n = 179), ми також обмежили всі інші набори даних 179 найбільш використовуваними ліками. Для кожної країни ми підрахували відсоток окремих 179 препаратів зі списку. На основі цих процентних співвідношень ми розрахували середній рейтинг у всіх шести країнах. Розрахунок списку 100 найкращих препаратів проводився за допомогою програмного забезпечення Stata Statistical Software: Release 16 (StataCorp College Station, Техас: StataCorp LLC, США).
З остаточного списку обрали 100 найбільш виписуваних препаратів і провели пошук літератури.
Стратегія пошуку
Два автори (DBI та NA) здійснили пошук літератури з січня по жовтень 2021 року за допомогою бази даних PubMed (Medline). Провели окремий пошук літератури щодо ферментів, що метаболізують ліки, і транспортерів ліків.
Для окремих препаратів були застосовані такі рядки пошуку:
Метаболізм препарату : (назва препарату І (печінковий АБО печінковий АБО кишковий АБО CYP АБО цитохром P450) І (фармакокінетичний АБО ADME АБО метаболізм АБО метаболізований) І in vitro).
Транспортер ліків : (назва препарату І субстрат І (транспортер АБО транспорт) І (елімінація АБО виведення АБО екскреція АБО витік АБО поглинання)).
Пошуковий термін «назва препарату» був визначений як назва препарату, зазначеного в списку 100 найкращих. PubMed автоматично включив синоніми до назви препарату, наприклад, пошук парацетамолу також включав ацетамінофен. Крім того, ми визначили та включили відповідні посилання зі статей, визначених за допомогою рядка пошуку. Автор включили статті незалежно від року публікації, але прагнули використовувати найновіші опубліковані.
Крім того, провели пошук літератури для кожного препарату, щоб визначити, чи був препарат класифікований як проліки.
Проліки визначалися як неактивна речовина, яку потрібно перетворити на фармакологічно активну речовину за допомогою метаболізму або фізико-хімічних процесів.
Відбір досліджень для 100 найпопулярніших препаратів
Перед пошуком літератури творили три критерії включення. Критерії включення полягали в тому, що статті, включені в аналіз, мали бути специфічними для відповідного препарату та мали повідомляти (i) вихідні дані, (ii) дослідження in vitro на людях та (iii) дані фази I або II метаболізму або транспортування ліків.
Документи Управління з харчових продуктів і медикаментів (FDA) або Європейського агентства з лікарських засобів (EMA) [наприклад, Коротка характеристика препарату (SmPC) або Основні відомості про призначення] були включені в аналіз, якщо вони були знайдені як посилання на статті під час пошуку літератури. Два автори (DBI та NA) відповідали за пошук 50 препаратів зі списку 100 найкращих і незалежно один від одного перевіряли відповідні статті, знайдені під час пошуку літератури. Автори не перевіряли всі статті, отримані під час пошуку літератури, а лише до тих пір, поки метаболізм і транспортування лікарського засобу не було підтверджено через прийнятну статтю, яка вважалася дійсною відповідно до критеріїв включення.
Аналіз даних
Для препарату, про який йде мова, позначали фермент або транспортер як головний, якщо була досягнута одна з двох умов: (i) якщо стаття з літературного пошуку стверджувала, що фермент або транспортер відповідають за понад 50% фракції, що метаболізується або транспортується або (ii) якщо в оригінальному документі фермент або транспортер визначено як «основний». Автори не визначили нижню межу для класифікації ферментів або транспортерів як другорядних на основі внеску в метаболізм або транспортування ліків. Крім того, реєстрували статус препарату (проліки чи ні). Автори розділили ферменти фази I на підкатегорії ізоформ, наприклад, CYP2C9 і CYP2D6, і підрахували кількість 100 препаратів, метаболізованих кожною ізоформою. Усі ізоформи зі списку Топ-100 були однаково включені в розрахунок незалежно від того, чи були вони позначені як головний фермент чи транспортер. Ізоформи CYP3A4 і CYP3A5 були згруповані в одну ізоформу, CYP3A4/5, оскільки вони мають структурну подібність на 80% і мають субстратну специфічність, що перекривається, що ускладнює розрізнення ізоформ.
Для ферментів фази II ми лише розділили уридин-5'-дифосфо-глюкуронозилтрансферази (UGT) на ізоформи, наприклад, UGT2B7 та UGT1A9, оскільки UGT є найбільшою надродиною фази II. Решта ферментів фази II були згруповані до надродини сульфотрансфераз (SULT). Для транспортерів ліків ми описали P-глікопротеїн (P-gp), білок резистентності раку молочної залози (BCRP) і поліпептид, що транспортує органічні аніони (OATP) в ізоформах, а також згрупований транспортер органічних аніонів (OAT), транспортер органічних катіонів (OCT), множинну лікарську стійкість ‐асоційований білок 1–4 (MRP1–4) і екструзія множинних лікарських і токсичних сполук (MATE) у суперродини. Ізоформи та суперродини як ферментів фази II, так і транспортерів ліків були згруповані як « Інші» якщо вони метаболізували або транспортували ≤2 зі 100 препаратів.
Для обох ферментів фази I та фази II та транспортерів ліків розраховували перекривання субстратів у кожній групі (фаза I, фаза II та транспортери ліків). Автори згрупували перекриття субстратів у такі групи для ферментів фази I: 1–5 препаратів, 6–10 препаратів і більше 10 препаратів. Для ферментів II фази та транспортерів ліків змінили діапазон накладення субстратів, щоб краще відображати перекривання субстратів у цих групах. Автори згрупували субстратне накладання як 1–2 препарати, 3–4 препарати та більше 4 препаратів. Також розрахували субстратне перекриття між транспортерами ліків (P-gp, BCRP і OATP1B1/3) і чотирма ферментами CYP (CYP3A4/5, CYP2D6, CYP2C9 і CYP2C19). Згрупували субстратне перекриття між транспортерами ліків і ферментами CYP як 1–5 препаратів, 6–10 препаратів і більше 10 препаратів.
РЕЗУЛЬТАТИ
Зі 100 препаратів, які найчастіше призначають, відомо, що 89 препаратів піддаються метаболізму або транспорту ліків, тоді як решта 11 препаратів не метаболізуються або є субстратами для переносників ліків. Ферменти фази I метаболізують 67 ліків, 18 ліків метаболізуються ферментами фази II, а відомо, що 54 препарати транспортуються транспортерами ліків. Відомо, що загалом 27 препаратів є субстратами як для фази I метаболізму, так і для транспорту ліків, тоді як відомо, що лише сім препаратів є субстратами для фази I та II метаболізму та транспорту ліків
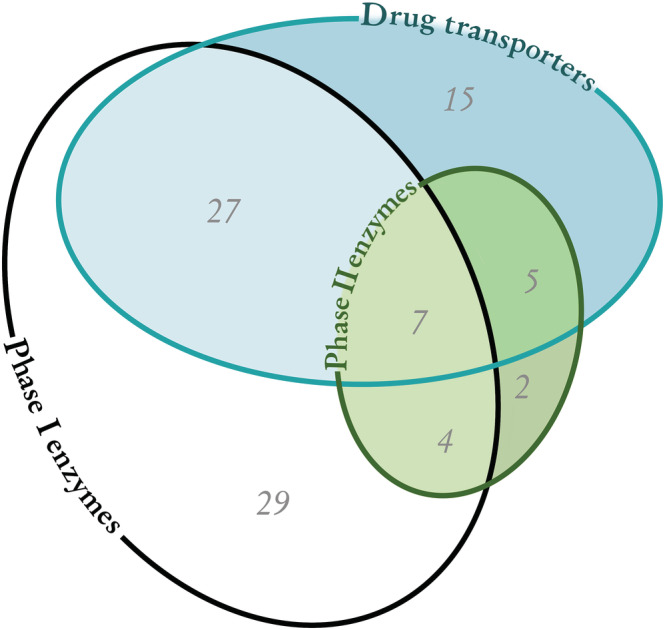
Кількість зі 100 препаратів, які найчастіше призначаються, метаболізуються ферментами фази I або ферментами фази II або транспортуються транспортерами ліків. Області, що перекриваються, відображають комбінацію шляхів. Зі 100 найбільш виписуваних препаратів 11 препаратів не метаболізуються та не транспортуються.
І фаза метаболізму
CYP3A4/5 є найважливішим ферментом фази I, оскільки він бере участь у метаболізмі 43 зі 100 препаратів, за ним йдуть CYP2D6 (23 препарати), CYP2C9 (23 препарати), CYP2C19 (22 препарати), CYP1A2 (14 препаратів) та CYP2C8 (11 препаратів).
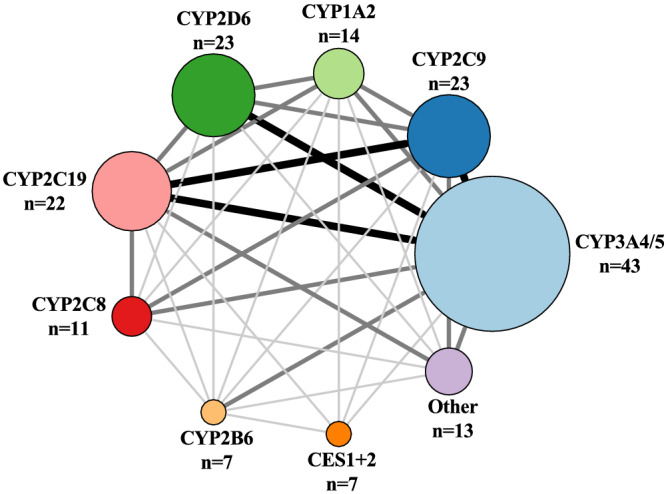
Кількість лікарських засобів, що метаболізуються ферментами фази I, ілюструється збільшенням розміру кола. Розмір і темність ліній між ферментами ілюструють перекривання субстратів між ферментами. Тонка/світло-сіра лінія відповідає 1–5 лікарським засобам у вигляді перекриття субстрату. Середня/сіра лінія відповідає 6–10 лікарським засобам як накладення субстрату. Товста/чорна лінія відповідає >10 лікарським засобам як накладення субстрату. Перекриття всередині групи не показано на малюнку, але ми звертаємося до таблиці для отримання додаткової інформації. Група Інше включає CYP1A1, CYP2E1, CP2C18, CYP2J2, CYP3A3, CYP3A7, CYP2A6, CYP1B1, AO, FMO та MAO-A + B. AO, карбоксилестераза; CYP, фермент цитохрому Р450; FMO, альдегідоксидаза; CES, флавін-вмісна монооксигеназа; МАО, моноаміноксидаза.
Виявили, що CYP3A4/5 метаболізує 43 зі 100 препаратів і має субстратне перекриття з трьома ізоформами CYP, CYP2D6, CYP2C9 та CYP2C19 (>10 препаратів для всіх ферментів)
CYP3A4/5 переважно експресується в печінці та кишечнику і є найбільш домінуючим ферментом, що метаболізує ліки в організмі. Топ-3 препарати з нашого списку з CYP3A4/5 як основний фермент включають бісопролол, етинілестрадіол і зопіклон.
CYP3A4/5 регулюється транскрипцією рецептором прегнану Х (PXR) і конститутивним рецептором андростану (CAR). PXR і CAR є членами надродини ядерних рецепторів (NR), і PXR є одним з найважливіших рецепторів у регуляції метаболізму та транспорту ліків. Флуклоксацилін (номер 61 зі 100 препаратів), диклоксацилін (номер 77) і рифампіцин (не в списку) є прикладами агоністів PXR, які викликають регуляцію CYP3A4 за допомогою PXR. Навпаки, ципрофлоксацин (номер 92) є прикладом помірного інгібітору CYP3A4. Час, потрібний для індукції або інгібування, і час, потрібний ферменту для відновлення після цього, є важливим при оптимізації фармакотерапії. В одному клінічному дослідженні досліджували час відновлення CYP3A4 після застосування рифампіцину протягом 7 днів і виявили, що для відновлення після опосередкованої рифампіцином індукції необхідно 8 днів. В іншому клінічному дослідженні досліджували індукцію CYP3A4 після 28 днів лікування рифампіцином. Для повного відновлення після індукції, опосередкованої рифампіцином, було необхідно припинити лікування на 28 днів. Тривалість конкурентного інгібування залежить від періоду напіввиведення інгібітору, тоді як тривалість неконкурентного інгібування та індукції залежить від різних біологічних факторів, таких як деіндукція транскрипції, керованої PXR (наприклад, для CYP3A4), деградація індукованих мРНК та їх кодовані білки в печінці та кишечнику, синтез білка CYP та обмін клітин.
Їжа, напої, рослинні препарати та запалення також можуть регулювати експресію CYP3A4/5 і призводити до концентрації препарату поза терапевтичним діапазоном. І севільський апельсин, і грейпфрут пригнічують CYP3A4. Звіробій, рослинний препарат, який використовується проти депресії, є потужним лігандом PXR та індуктором CYP3A4. CYP3A4/5 також може знижуватися прозапальними цитокінами під час запалення.
Незважаючи на численні дослідження генетичного поліморфізму CYP3A4, наразі немає доказів поширених і клінічно значущих поліморфізмів CYP3A4. Особи з генетичним поліморфізмом CYP3A5 можуть бути охарактеризовані як експресори CYP3A5 або як неекспресори CYP3A5. Неекспресори CYP3A5 складають 80–85% європеоїдів, тому експресори CYP3A5 є меншістю в Європі. Це на відміну від азіатського та афроамериканського населення, де 60%–73% і 32% не є експресорами CYP3A5 відповідно. Рекомендації Консорціуму впровадження клінічної фармакогенетики (CPIC) для лікування такролімусом (субстрат CYP3A4/5) (немає в списку) рекомендують стандартну початкову дозу для неекспресора CYP3A5 та вищу початкову дозу для експресорів CYP3A5. Результати показують, що CYP2D6 метаболізує 23 зі 100 препаратів і має найбільше субстратне перекриття з CYP3A4/5 (10 препаратів). CYP2D6 в основному експресується в печінці. Це другий за важливістю фермент CYP у цьому огляді, і на його частку припадає 5% загального вмісту білка CYP у печінці людини. Топ-3 препарати з нашого списку з CYP2D6 як основний фермент включають метопролол, венлафаксин і флуоксетин.
CYP2D6 не сприйнятливий до індукції ферменту ліками, але експресія CYP2D6 підвищується під час вагітності. У клінічному дослідженні плазмовий метаболічний коефіцієнт декстрометорфану, досліджуваного CYP2D6 (не в списку), знизився на 53% під час вагітності порівняно з періодом після вагітності. Антидепресанти сертралін (номер 19) і флуоксетин (номер 52) є інгібіторами CYP2D6. Інгібування триває 5 днів для сертраліну та 42 дні для флуоксетину після припинення. Лікарі повинні бути обережними, призначаючи субстрати CYP2D6 після початку та припинення прийому цих антидепресантів.
Поліморфізм ферментів CYP відіграє вирішальну роль, особливо CYP2D6. Мета-аналіз показав, що ефективність метопрололу (номер 16) є вищою у пацієнтів зі слабким метаболізмом, ніж у тих, хто не має поганого метаболізму. Рекомендації CPIC рекомендують альтернативні анальгетики двом пролікам, кодеїну (номер 7) і трамадолу (номер 21), у тих, хто повільно або надшвидко метаболізує, оскільки вони, швидше за все, відчуватимуть незначне полегшення болю або більше побічних ефектів порівняно з особами з проміжним метаболізмом. Нещодавно було виявлено, що ядерний фактор 1B (NFIB) регулює експресію гена CYP2D6 in vitro. Те саме дослідження показало, що носії NFIB rs28379954 T>C, які також були Особи з інтенсивним метаболізмом CYP2D6 мали порівнянну активність CYP2D6 з тими, хто надшвидко метаболізував. Це підкреслює, що поліморфізм NFIB важливо враховувати при метаболізмі ліків CYP2D6.
Автори показують, що CYP2C9 метаболізує 23 зі 100 препаратів і має субстратне перекриття з CYP3A4/5 і CYP2C19 (>10 препаратів для кожного ферменту). CYP2C9 переважно експресується в печінці та шлунково-кишковому тракті. Сімейство CYP2C містить CYP2C8, CYP2C9 і CYP2C19, які загалом складають 33% від загального вмісту білка CYP у печінці, при цьому CYP2C9 становить 24%. Топ-3 препарати з нашого списку з CYP2C9 як основний фермент включають напроксен, диклофенак і варфарин. CYP2C9 регулюється трьома ядерними рецепторами: PXR, CAR і глюкокортикоїдним рецептором (GR).
S-варфарин (номер 45) є субстратом CYP2C9 з вузьким терапевтичним індексом. Флуконазол (номер 87) інгібує CYP2C9, і епідеміологічне дослідження показало, що одночасне застосування флуконазолу з варфарином призводить до підвищення середнього міжнародного нормалізованого відношення (INR) на 0,83. Це клінічно значуще збільшення INR може призвести до побічних ефектів. Диклоксацилін (номер 77) є індуктором CYP2C9 через активацію PXR. Два епідеміологічні дослідження показали, що одночасне застосування диклоксациліну та варфарину призводить до зниження середнього МНВ (INR) на 0,62 після 2–4 тижнів застосування диклоксациліну та підвищення ризику ішемічного інсульту та системної емболії (коефіцієнт ризику 2,19).
Генетичний поліморфізм може впливати на активність CYP2C9. CYP2C9*2 і CYP2C9*3 є найбільш вивченими генотипами, носії обох мають знижену активність CYP2C9. Плазмовий кліренс S-варфарину знижується на 56% (CYP2C9*1/3), 70% (CYP2C9*2/3) і 75% (CYP2C9*3/3) порівняно з диким типом (CYP2C9*1/1). Три великі рандомізовані клінічні випробування досліджували, чи генотипування перед початком антикоагулянтної терапії покращує відсоток часу в терапевтичному діапазоні INR. Дослідження дійшли суперечливих висновків, що ускладнило впровадження варфарину під контролем CYP2C9. Рекомендації CPIC щодо терапії варфарином рекомендують дозування на основі генотипу, лише якщо він відомий до початку лікування.
CYP2C19 метаболізує 22 зі 100 лікарських засобів і має значне перекриття субстратів зі CYP2C9 та CYP3A4/5 (>10 препаратів для кожного ферменту). CYP2C19 експресується в печінці та шлунково-кишковому тракті. Топ-3 препарати з нашого списку з CYP2C19 як основний фермент включають омепразол, пантопразол та езомепразол. CYP2C19 регулюється тими ж трьома ядерними рецепторами, що й CYP2C9 (PXR, CAR і GR). Рифампіцин (не в списку) є індуктором цього ферменту через CAR і PXR, а дексаметазон (номер 60) індукує CYP2C19 через GR.
Поліморфізм CYP2C19 був широко досліджений, і деякі варіанти можуть бути клінічно значущими. Носії CYP2C19*17 характеризуються як надшвидкі метаболізатори, тоді як носії CYP2C19*2/3 характеризуються як повільні метаболізатори. Пацієнти, які є носіями CYP2C19*2 і які лікуються циталопрамом (номер 30), субстратом CYP2C19, мали нижчу ймовірність толерантності до препарату. Клінічне дослідження показало, що люди, які є носіями CYP2C19*2 і CYP2C19*3, мали нижчий метаболізм омепразолу (номер 4) порівняно з особами, які є носіями CYP2C9*1. Це ж дослідження показало, що прийом інгібітору CYP2C19 флувоксаміну (не в списку) знижує метаболізм омепразолу в осіб, які є носіями CYP2C19*1, але не впливає на метаболізм омепразолу у носіїв CYP2C19*2 і CYP2C19*3 проміжних і повільних метаболізаторів CYP2C19, які отримують проліки клопідогрелю (номер 36), відчувають знижене пригнічення тромбоцитів і підвищений ризик серйозних несприятливих серцево-судинних і цереброваскулярних подій. Настанова CPIC рекомендує розглянути альтернативу клопідогрелю для проміжних метаболізаторів і уникати клопідогрелю для повільних метаболізаторів.
Огляд показує, що CYP1A2 метаболізує 14 зі 100 препаратів і має субстратне перекриття з CYP3A4/5, CYP2C9, CYP2C19 і CYP2D6 (6–10 препаратів для кожного ферменту). CYP1A2 переважно експресується в печінці. З списку CYP1A2 не класифікується як основний фермент у метаболізмі ліків, але трьома найкращими субстратами CYP1A2 зі списку є напроксен, етинілестрадіол і міртазапін. CYP1A2 регулюється транскрипцією арил-вуглеводневим рецептором (AhR), який є ліганд-активованим фактором транскрипції.
Деякі препарати індукують CYP1A2. Омепразол (номер 4) є індуктором CYP1A2 in vitro. Найпотужніші інгібітори CYP1A2 є плоскими молекулами з невеликим об’ємом, які легко вписуються в активний центр CYP1A2. Вони часто містять метильні, хлорні або фторні замінники, наприклад, ципрофлоксацин (номер 92). Ципрофлоксацин і оральні контрацептиви, що містять етинілестрадіол (номер 27) і гестоген, були досліджені на пригнічення метаболізму тизанідину (субстрат CYP1A2) (не в списку). Ципрофлоксацин є сильнішим інгібітором, ніж оральні контрацептиви; однак слід бути обережним при застосуванні тизанідину особам, які використовують оральні контрацептиви, оскільки тизанідин має вузький терапевтичний діапазон.
Активність CYP1A2 залежить від індивідуальних відмінностей від генетичних факторів і факторів навколишнього середовища, таких як куріння. Клозапін (не в списку) є субстратом CYP1A2 і є антипсихотичним препаратом, де використовується терапевтичний моніторинг препаратів. Мета-аналіз рекомендував зменшити дозу клозапіну на 30% для пацієнтів, які палять і раптово припиняють палити, а також проаналізувати рівень клозапіну в крові.
Автори виявили, що CYP2C8 метаболізує 11 зі 100 препаратів і має субстратне перекриття з CYP3A4/5, CYP2C9 і CYP2C19 (6–10 препаратів для кожного ферменту). CYP2C8 сильно експресується в печінці. З нашого списку CYP2C8 не класифікується як основний фермент у метаболізмі 100 найбільш використовуваних ліків, але трьома найпопулярнішими субстратами CYP2C8 з нашого списку є ібупрофен, етинілестрадіол і зопіклон. Регуляція транскрипції CYP2C8 така ж, як і для CYP2C9 і CYP2C19 (PXR, CAR і GR).
Фелодипін (номер 76) є прикладом потужного інгібітору in vitro, а триметоприм (номер 55) є слабким інгібітором CYP2C8 як in vitro, так і in vivo. Клопідогрель (номер 36) є інгібітором CYP2C8 через його метаболіт, ацил-β-D-глюкуронід клопідогрелю. Ретроспективне дослідження показало, що пацієнти, які отримували паклітаксел (субстрат CYP2C8) (немає в списку), мали приблизно 2-кратне підвищення ризику розвитку нейропатії 2 ступеня або вище при одночасному лікуванні клопідогрелем. Виявлено лише кілька індукторів CYP2C8. Дексаметазон (номер 60), кортикостероїд, індукує CYP2C8 шляхом зв’язування з GR. Рифампіцин (не в списку) індукує CYP2C8 і збільшує експресію ферменту через активацію PXR.
Генетичний поліморфізм CYP2C8*3 є найбільш дослідженим поліморфізмом CYP2C8. Цей алель поширений у європеоїдів, але рідко зустрічається в популяціях Африки та Азії. Багато досліджень вивчали цей алель, але дані суперечливі щодо впливу на метаболізм, і активність CYP2C8*3 може залежати від субстрату.
II фаза метаболізму
Суперродина UGT є найважливішою суперродиною фази II і бере участь у метаболізмі 17 найбільш використовуваних ліків, за якою йде сульфотрансфераза (SULT), яка метаболізує третину зі 100 ліків
Надродину UGT можна розділити на чотири родини: UGT1, UGT2, UGT3 і UGT8; однак UGT3 і UGT8 не мають істотного значення в метаболізмі ліків. Ми виділяємо UGT1A та UGT2B як найважливіші члени родини для метаболізму ліків. Ми показали, що UGT2B7 відповідає за метаболізм дев’яти зі 100 ліків). UGT1A3 і UGT1A9 є другими за важливістю ферментами фази II, і вони метаболізують вісім зі 100 ліків). UGT2B7 і UGT1A9 мають найбільше перекриття субстрату з п'ятьма препаратами. UGT в основному експресуються в печінці та кишечнику.
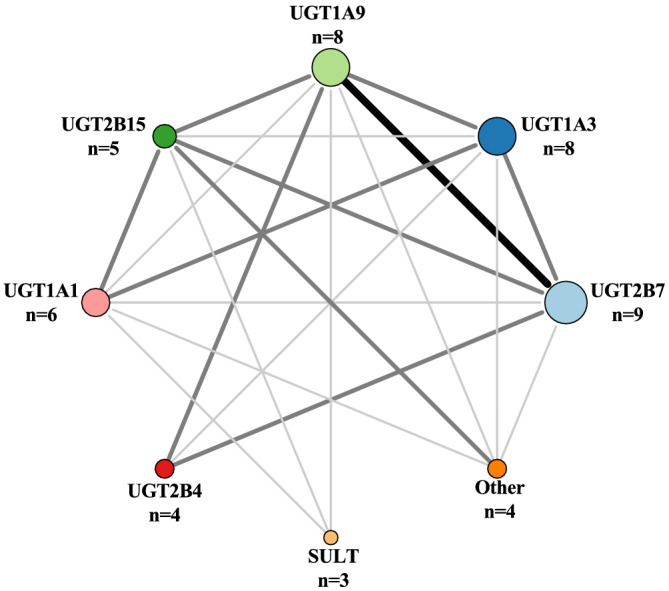
Кількість лікарських засобів, що метаболізуються ферментами фази II, ілюструється збільшенням розміру кола. Розмір і темність ліній між ферментами ілюструють перекриття субстратів. Тонка/світло-сіра лінія відповідає 1–2 препаратам у вигляді накладання субстрату. Середня/сіра лінія відповідає 3–4 препаратам у вигляді накладання субстрату. Товста/чорна лінія відповідає >4 препаратам як накладення субстрату. Перекриття всередині групи не показано на малюнку, але ми звертаємося до таблиці для отримання додаткової інформації. Група Інше містить UGT1A4, UGT1A6 і UGT2B10. SULT, сульфотрансферази; UGT, уридин 5'-дифосфо-глюкуронозилтрансферази.
UGT позначено як основний фермент для двох зі 100 найкращих препаратів, що включає диклофенак і телмісартан. Різні ядерні рецептори беруть участь у регуляції різних ізоформ у надродині UGT. Це включає AhR, CAR, PXR, фарнезоїдний X-рецептор (FXR), печінковий X-рецептор (LXR) і рецептор, активований проліфератором пероксисом (PPAR). Досліджень, присвячених регулюванню UGTs, мало порівняно з ферментами CYP. Однак деякі препарати були ідентифіковані як інгібітори UGTs in vitro, хоча деякі з них підтверджені дослідженнями in vivo. UGT1A1 бере участь у глюкуронізації білірубіну, і дослідження показало, що інгібітори тирозинкінази пригнічують UGT1A1, що підвищує ризик гіпербілірубінемії у пацієнтів. Атазанавір (не в списку) використовується для лікування ВІЛ і інгібує UGT1A1. Якщо генотип UGT1A1 відомий до початку лікування, рекомендації CPIC рекомендують розглянути альтернативу атазанавіру препарату для пацієнтів з повільним метаболізмом, оскільки існує підвищений ризик жовтяниці. Дослідження in vitro також показали, що UGT підлягають індукції. Багато індукторів UGT також індукують інші ферменти, такі як ферменти CYP, і включають рифампіцин (немає в списку), фенобарбітал (немає в списку) і карбамазепін (немає в списку). Дослідження in vivo з вивчення індукції або інгібування важко провести, оскільки існує небагато хороших і специфічних пробних препаратів для ізоформ UGT.
P-gp є найважливішим транспортером ліків і, як відомо, транспортує 30 зі 100 ліків. Відомо, що білок резистентності до раку молочної залози (BCRP) транспортує 11 ліків, а поліпептид 1B1 (OATP1B1) і OATP1B3, що транспортують органічні аніони, транспортують дев’ять ліків.
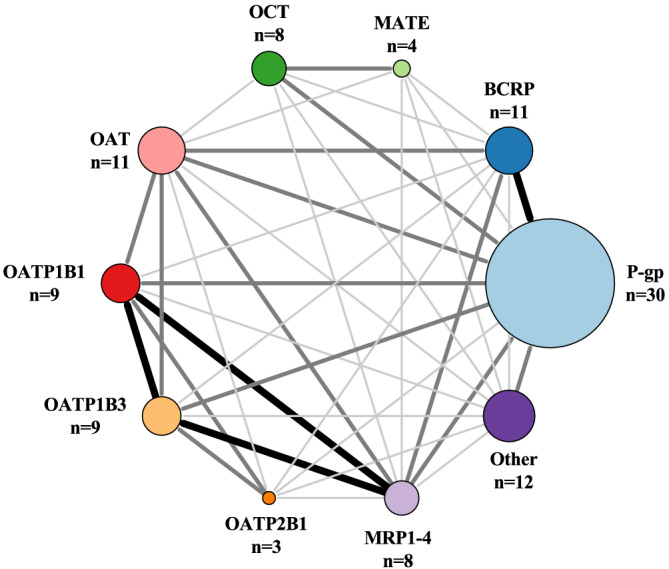
Кількість наркотиків, що перевозяться транспортними засобами, показано зі збільшенням розміру кола. Розмір і темність ліній між ферментами ілюструють перекриття субстратів. Тонка/світло-сіра лінія відповідає 1–2 препаратам у вигляді накладання субстрату. Середня/сіра лінія відповідає 3–4 препаратам у вигляді накладання субстрату. Товста/чорна лінія відповідає >4 препаратам як накладення субстрату. Перекриття всередині групи не показано на малюнку, але ми посилаємося на таблицю для подальшої інформації. Група Інше містить MCT, OATP1A2, OATP4C1, OCTN, LAT, SERT, PMAT, THTR, CHT, NTCP, PePT і AE. АЕ, аніонообмінний білок; BCRP, білок стійкості до раку молочної залози; CHT, транспортер холіну; LAT, транспортер L-амінокислот; MATE, екструзія з кількома препаратами та токсичними сполуками; MCT, транспортер монокарбоксилату; MRP, білок, пов’язаний з множинною резистентністю; NTC, співтранспортуючий поліпептид таурохолат натрію; OAT, переносник органічних аніонів; OATP, поліпептид, що транспортує органічні аніони; OCT, переносник органічних катіонів; OCTN, роман про переносник органічних катіонів; PePT, транспортер пептидів; PMAT, транспортер моноамінів плазматичної мембрани; P-gp, P-глікопротеїн; SERT, транспортер серотоніну; THTR, білок-транспортер тіаміну.
Автори виявили що відомо, що P-gp транспортує 30 ліків і має найбільше субстратне перекриття з BCRP (п’ять препаратів). Субстратне перекриття 3-4 препаратів спільно з OATP1B1 і OATP1B3, транспортером органічних катіонів (OCT), транспортером органічних аніонів (OAT) і білком множинної лікарської стійкості 1-4 (MRP1-4). Найбільше загальне перекриття субстратів для P-gp відбувається з CYP3A4/5 (>10 препаратів), і незначне накладення субстрату спостерігається з CYP2D6, CYP2C9 та CYP2C19 (6–10 препаратів).
P-gp має широке поширення в тканинах, наприклад, головному мозку, ендокринних тканинах, шлунково-кишковому тракті, печінці та нирках. Він належить до надродини транспортерів АТФ-зв’язувальної касети (ABC), а функція P-gp полягає в обмеженні клітинного накопичення ендогенних метаболітів і ксенобіотиків.
З нашого списку P-gp не класифікується як основний транспортер у транспортуванні ліків зі 100 найбільш використовуваних ліків, але 3 найкращі субстрати P-gp з нашого списку включають аторвастатин, омепразол і лозартан. P-gp кодується геном полірезистентності людини (MDR1), який регулюється двома рецепторами, PXR і CAR.
Рифампіцин (не в списку) є добре відомим індуктором P-gp, і його вплив на P-gp широко вивчається з різними субстратами, наприклад, фексофенадином (номер 84). Клінічне дослідження показало, що рифампіцин зменшує площу під кривою (AUC) фексофенадину на 51%. Верапаміл (не в списку) є добре відомим інгібітором P-gp, і одночасне застосування з фексофенадином призводить до 2,5-кратного збільшення AUC фексофенадину у чоловіків-добровольців.
Генетичні поліморфізми в MDR1 потенційно можуть змінити функціональну експресію та активність, але, незважаючи на широкі дослідження в цій галузі, досі немає консенсусу щодо значення генетичного поліморфізму P-gp.
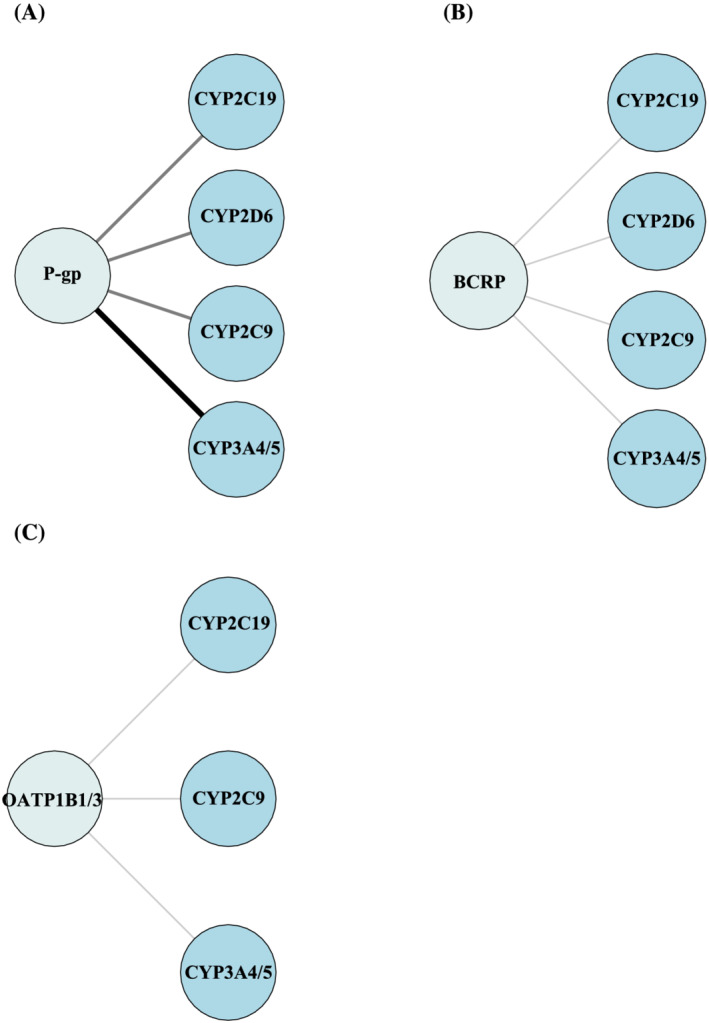
Білок стійкості до раку молочної залози (BCRP)
Огляд показує, що відомо, що BCRP транспортує 11 лікарських засобів, і, окрім субстратного перекриття п’яти препаратів із P-gp, BCRP має спільні субстрати 3–4 препаратів з OAT та MRP1–4. Зі списку, BCRP не класифікується як основний транспортер у транспортуванні наркотиків зі 100 найбільш використовуваних ліків, але 3 найпопулярніші субстрати BCRP у списку – пантопразол, фуросемід і розувастатин.
BCRP знаходиться, наприклад, у мозку, шлунково-кишковому тракті, репродуктивних органах і м'язових тканинах. Основний субстрат BCRP з нашого списку включає алопуринол (номер 51). BCRP належить до надродини транспортерів ABC, а транспортер BCRP кодується геном ABCG2.BCRP регулюється AhR, CAR, PXR, GR, рецептором естрогену β (ER-β), PPAR-γ і фактором 2, пов’язаним з ядерним фактором, еритроїдним 2 (Nrf2).
Фебуксостат (немає в списку) є новішим інгібітором ксантиноксидази, який пригнічує BCRP-опосередкований транспорт розувастатину (номер 24) як in vitro, так і in vivo. Ген ABCG2 є поліморфним, а варіант ABCG2 c.421C>A добре вивчений. Мінорний алель А призводить до зниження експресії білка BCRP на 30–40% порівняно з еталонним алелем. Дослідження показало, що варіант ABCG2 c.421C>A призводить до поганої відповіді на субстрат BCRP алопуринол. Фармакокінетичні дані показують, що пацієнти з варіантом ABCG2 c.421C>A мають підвищену експозицію розувастатину (на 144% збільшення AUC), що призводить до більш високого ризику міопатії. Крім того, загальногеномні асоційовані дослідження показали, що носії варіанту мають покращену реакцію розувастатину на зниження рівня холестерину. На основі цих результатів настанова CPIC рекомендує пацієнтам з поганою функцією ABCG2 зменшити початкову дозу розувастатину, що є субстратом BCRP, до ≤20 мг або розглянути альтернативні статини, якщо потрібно більше 20 мг.
Відомо, що OATP1B1 і OATP1B3 транспортують дев’ять наркотиків. Найбільше субстратне перекриття для двох ізоформ відбувається одна з одною (вісім препаратів) і з MRP1–4 (п’ять препаратів). OATP1B1 і OATP1B3 також мають субстратне перекривання трьох препаратів з OAT та перекриття субстрату з CYP3A4/5, CYP2C9 та CYP2C19 (1–5 препаратів). OATP1B1 і OATP1B3 переважно експресуються в печінці, і мають 80% амінокислотної гомології. З нашого списку OATP1B1 і OATP1B3 не класифікуються як головні транспортери в транспорті ліків 100 найбільш використовуваних ліків, але 3 найкращі субстрати OATP1B1 і OATP1B3 з нашого списку - це аторвастатин, симвастатин і фуросемід. OATP1B1 і OATP1B3 є членами сімейства переносників розчинених речовин (SLC), які регулюють поглинання клітинами і кодуються SLCO1B1 (OATP1B1) і SLCO1B3 (OATP1B3). Обидва транспортери функціонують як активні транспортери поглинання. Регуляція транскрипції OATP1B1 і OATP1B3 відрізняється. Основними регуляторами транскрипції для OATP1B1 є FXR і LXRα, але відомо, що тільки FXR бере участь у регуляції OATP1B3.
Рифампіцин (не в списку) є інгібітором OATP1B1 і OATP1B3. Дослідження за участю здорових добровольців дало учасникам дослідження одну дозу рифампіцину та показало семикратне збільшення AUC аторвастатину (номер 2), субстрату OATP1B1 та OATP1B3.
Генотип c.521T>C у SLCO1B1 є найбільш широко вивченим поліморфізмом. Цей варіант призводить до зниження транспортної активності in vitro. Декілька досліджень показали, що носії генотипу c.521T>C мають підвищений ризик міотоксичності, пов’язаної зі симвастатином (номер 5). CPIC зазначено, що пацієнти зі зниженою або поганою функцією OATP1B1 мають підвищений ризик розвитку міопатії при лікуванні аторвастатином і симвастатином, і дозу слід відповідно відкоригувати. Наразі не існує добре підтвердженого поліморфізму в OATP1B3, який потребував би зміненої фармакотерапії.
Обговорення
У цьому огляді ми визначили 100 ліків, які найчастіше призначають у п’яти європейських країнах та Австралії. Ми виявили, що 89 зі 100 ліків метаболізуються або метаболізмом фази I, або метаболізмом фази II, або, як відомо, є субстратами для транспортувальників ліків, тоді як 11 препаратів не піддаються метаболізму або транспорту ліків. Загалом 67 зі 100 препаратів проходять фазу I метаболізму, де CYP3A4/5 є домінуючим ферментом, за яким йдуть CYP2D6, CYP2C9, CYP2C19, CYP1A2 та CYP2C8. UGT є найбільш домінуючими ферментами фази II, відповідальними за метаболізм 17 зі 100 ліків. Нарешті, P-gp є домінуючим транспортером і, як відомо, відповідає за транспортування 30 зі 100 ліків. CYP3A4/5 і P-gp мають значне перекриття субстратів 15 препаратів, і якщо експресія або активність змінюється для CYP3A4/5 або P-gp, це потенційно вплине на 73 препарати.
Виявлено, що внесок CYP3A4/5 у метаболізм препарату становив 30% і, отже, нижчий порівняно з попереднім дослідженням (37%) і поточним (43%). Під час пошуку літератури оцінювали метаболізм кожного препарату. Для проліків, активованих шляхом метаболізму, метаболізм лікарського засобу охоплює як шлях активації, так і шлях виведення прийнятого препарату. За будь-який зі цих шляхів відповідають різні ферменти, і це важливо враховувати, оскільки зміни в одному з цих ферментів по-різному впливатимуть на ефективність. Слід зазначити, що фармакотерапія постійно розвивається, особливо щодо ферментів II фази та транспортерів ліків, і, таким чином, постійно з’являються нові знання. Крім того, нові препарати схвалюються, а інші застарівають. Тому важливо оновлювати цей список у майбутньому. Сильні сторони цього огляду полягають у тому, що отримано дані про ліки, що відпускаються за рецептом, з п’яти країн Європи та Австралії, що робить список застосовним для багатьох країн. Крім того, використовували оригінальну літературу, засновану на дослідженнях людини in vitro. Обирали дослідження in vitro на людях, оскільки ці дослідження є найточнішими для пояснення основних механізмів метаболізму та транспорту ліків.
Висновок
На завершення було виявлено, що 89 зі 100 препаратів, які найчастіше призначають, метаболізуються та/або, як відомо, транспортуються. Лише 11 препаратів не піддаються ні метаболізму, ні транспорту ліків. Оскільки участь і перекривання ферментів і транспортерів є високими, це дослідження підкреслює ризик взаємодії між ліками у пацієнтів, які приймають кілька ліків. Таким чином, розуміння мінливості метаболізму та транспорту ліків залишається пріоритетним.
ДЖЕРЕЛО: https://www.ncbi.nlm.nih.gov/

Щоб дати відповіді на запитання до цього матеріалу та отримати бали,
будь ласка, зареєструйтеся або увійдіть як користувач.
Реєстрація
Вхід
Матеріали з розділу

Верховна Рада ухвалила законопроєкт 6364: ...

Дитяча травма, пов’язана з головним болем ...

У селах та на прифронтових територіях з'яв ...
Що відомо про новий штам SARS-CoV-2, який ...

Під час пандемії Covid більше людей захвор ...

Нові можливості оформлення згоди на посмер ...
